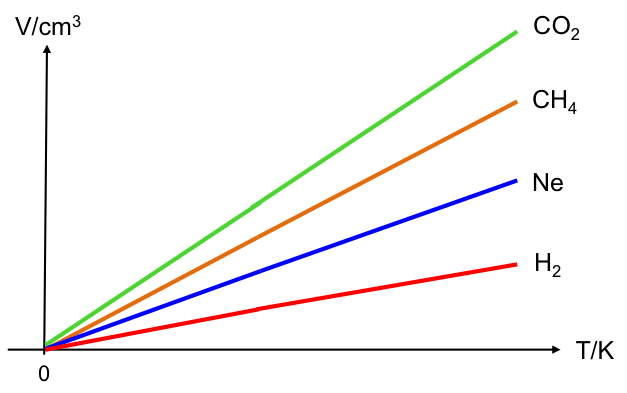
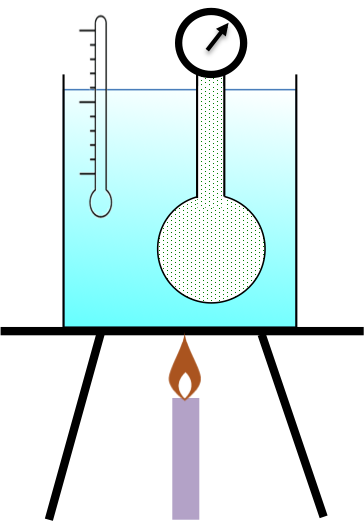
Measuring the change in enthalpy of a chemical reaction provides essential insights into the heat energy absorbed or released during the reaction.
The diagram below shows two devices for measuring enthalpy changes of reactions.
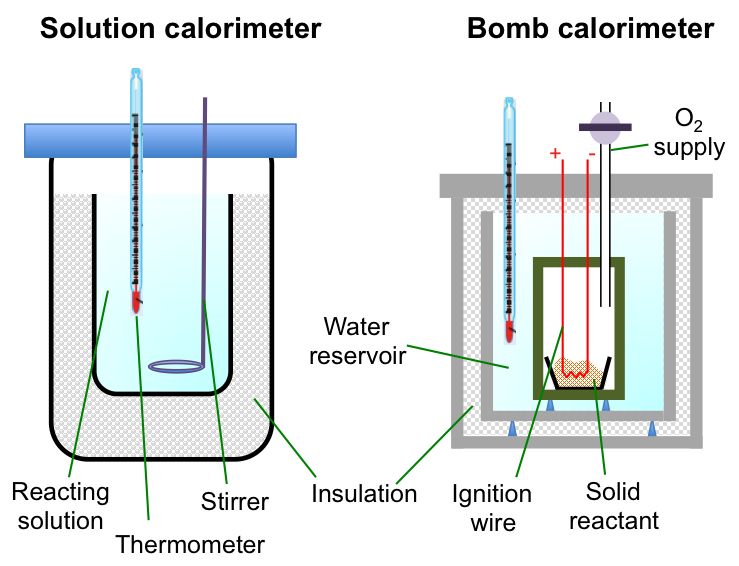
A calorimeter allows the change in temperature of the surroundings of a reaction system to be measured, and hence, the change in enthalpy of the reaction*.
The bomb calorimeter is useful for measuring the change in enthalpy of combustion reactions, e.g.
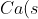+\frac{1}{2}O_2(g)\rightarrow&space;CaO(s))
The sample is placed in a crucible enclosed in a chamber made of a thermally conductive material. Oxygen is pumped into the chamber and the reaction is initiated by a current flowing through a wire embedded in the sample. All the energy released by the combustion reaction is assumed to flow across the chamber walls to heat up the water in the reservoir (surroundings). The change in enthalpy of the reaction is calculated by noting the change in temperature of the water reservoir. For example,
Mass of Ca used: 0.001000 kg
Mass of water in the reservoir: 0.4500 kg
Temperature change recorded: 13.10 K
Specific heat capacity of water: 4186 J K-1 kg-1
Using eq5, the enthalpy change for the reaction is:
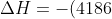(0.4500)(13.10)=-24676\:&space;J\approx-24700\:&space;J)
We have further assumed that heat gained by the chamber walls, and heat lost to the atmosphere, are negligible. The enthalpy change for one mole of calcium is:

Therefore,
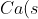+\frac{1}{2}O_2(g)\rightarrow&space;CaO(s)\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\Delta&space;H=-990\:&space;kJmol^{-1})
It is important to note that the enthalpy change quoted for a reaction changes when we modify the stoichiometric coefficients of the equation. For instance, the quoted ΔH doubles if we rewrite the above equation as:
+O_2(g)\rightarrow&space;2CaO(s)\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\Delta&space;H=-1980\:&space;kJmol^{-1})
In other words, we interpret ΔH = -990 kJmol-1 as the enthalpy change per one mole of calcium or half a mole of oxygen or one mole of calcium oxide for the reaction . Similarly, ΔH = -1980 kJmol-1 is the enthalpy change for two moles of calcium or one mole of oxygen or two moles of calcium oxide in the reaction +O_2(g)\rightarrow&space;2CaO(s)) .
.
Another perhaps less confusing way to quote the enthalpy change of a reaction is to drop the ‘per mole’ part of the unit to give:
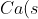+\frac{1}{2}O_2(g)\rightarrow&space;CaO(s)\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\Delta&space;H=-990\:&space;kJ)
+O_2(g)\rightarrow&space;2CaO(s)\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\Delta&space;H=-1980\:&space;kJ)
We use a solution calorimeter (see above diagram) when we want to measure the enthalpy change of an aqueous reaction, e.g. the neutralisation of hydrochloric acid with sodium hydroxide:
+NaOH(aq)\rightarrow&space;NaCl(aq)+H_2O(l))
Consider the reaction of 20.0 cm3 of 1.000 M HCl with 30.0 cm3 of 1.000 M NaOH resulting in an increase in temperature of 5.46 K. Note that water is considered the surroundings in this experiment, i.e. the reactants are dissolved in their surroundings. There are 0.0200 moles of HCl in 20.0 cm3 of water and 0.0300 moles of NaOH in 30.0 cm3 of water, which means that the total volume of solution is 50.0 cm3. Assuming that the density of the solution is the same as the density of water (1.00 g cm-3), the mass of the solution is 50.0 g. If we make the further assumption that the specific heat capacity of the solution is the same as that of water (4.18 Jg-1K-1), the change in enthalpy of the reaction is:
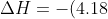(50.0)(5.46)=-1141\:&space;J\approx-1100\:&space;J)
Since HCl is the limiting reagent, the enthalpy change per mole of H2O is:

+OH^-(aq)\rightarrow&space;H_2O(l)\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\Delta&space;H=-57.1\:&space;kJmol^{-1})
* The bomb calorimeter actually measures the change in internal energy of the system, which we approximate it to be equal to the change of enthalpy. This is because the system in the bomb calorimeter is a constant-volume environment instead of a constant-pressure environment.