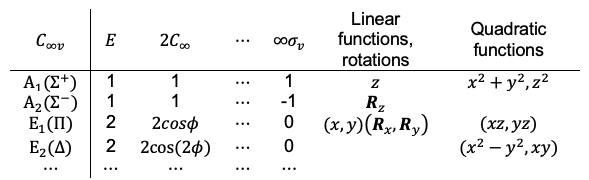
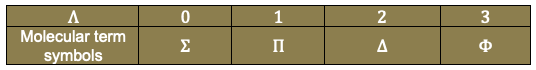
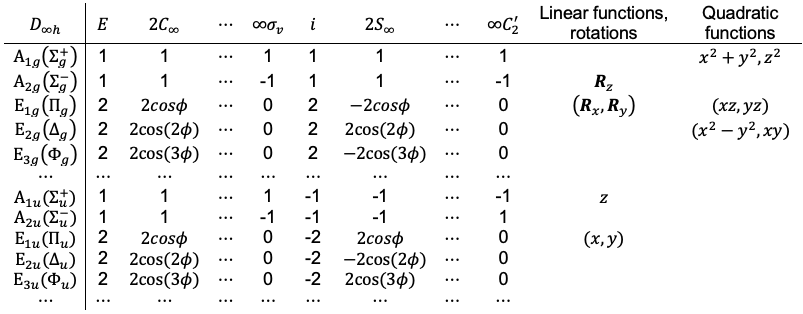
Maxwell’s fourth equation is a correction to Ampère’s law, stating that magnetic fields circulate around both electric currents and time-varying electric fields.
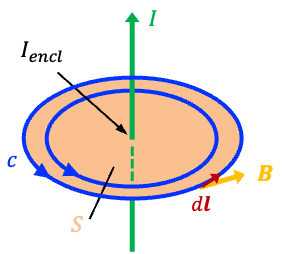
To understand why this correction was necessary, we first consider Ampère’s original law. From his experiments, Ampère observed that the circulation of the magnetic field around a closed loop
is proportional to the electric current
passing through the surface
enclosed by the loop (see diagram above). This relationship is expressed mathematically by the line integral
where is an infinitesimal vector line element tangent to the closed path
, whose magnitude is the differential path length and whose direction follows the chosen direction of integration around the loop, and
is the proportionality constant known as the permeability of free space.

Question
Why does a current flowing through a wire generate a magnetic field?
Answer
See this article for explanation.
Eq30 is the integral form of Ampère’s law. While it accurately describes steady currents, it becomes inconsistent for situations involving time-varying electric fields, such as a charging capacitor.
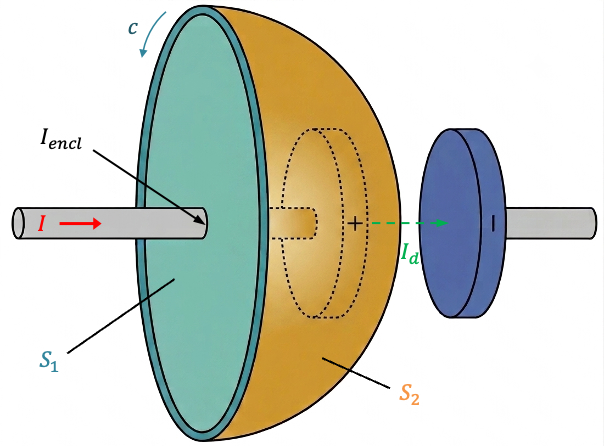
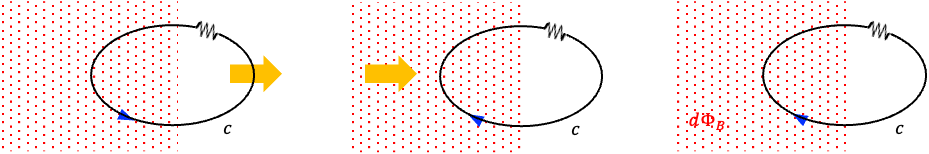
Consider a flat disc bounded by the closed loop
. This surface intersects the wire perpendicularly to the left of the positive plate of the charging capacitor and is pierced by the conducting current
(see diagram above). Therefore,
according to Ampère’s law. A second dome-shaped surface
, bounded by the same closed loop
, bulges out between the capacitor plates. It passes between the plates but does not intersect the conducting wire. For this surface,
, and Ampère’s law would predict
. However, the magnetic field surrounding the charging current does not disappear in the region between the capacitor plates. A magnetic needle placed near the gap still shows the same deflection as if it were placed near the conducting wire, indicating the presence of a magnetic field even though no conduction current flows across the gap. This contradicts the prediction of Ampère’s law.
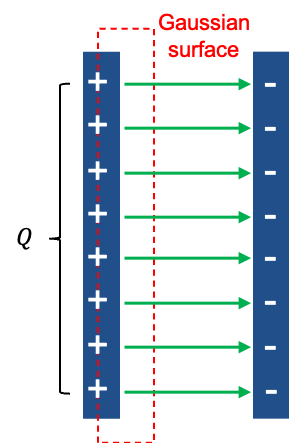
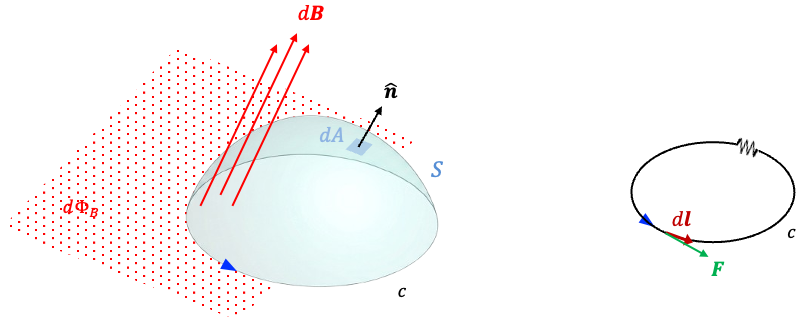
Maxwell resolved this inconsistency by introducing an effective current known as the displacement current flowing between the plates. To relate the electric field between the plates to the charge
on either plate, we first note that joining the surfaces
and
forms a closed surface. This closed surface encloses part of the conducting wire as well as the positive capacitor plate, so it is not convenient for relating the electric field in the gap directly to the charge on the plate. Instead, we consider a new Gaussian surface that encloses only the charge on the inner surface of the positive capacitor plate (see diagram above). The left face of this Gaussian surface lies just inside the conducting plate, where
, while its right face lies in the gap between the plates. Consequently, no electric flux passes through left face (
), or the top and bottom faces (assuming fringing fields are negligible), and all the electric flux passes through the right face towards the negative plate.
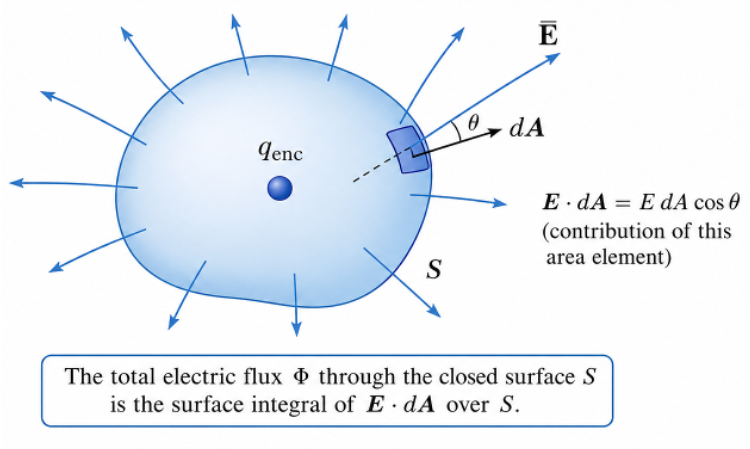
According to Gauss’s law, the electric flux through the Gaussian surface is . Differentiating this with respect to time gives:
Maxwell therefore defined the displacement current as . This leads to the integral form of Maxwell’s fourth equation (Ampère-Maxwell law):
For the surface , the enclosed current is entirely conduction current, so
. For
, no conduction current passes through the surface:
. Thus both surfaces yield the same value of
, removing the inconsistency in Ampère’s original law.
We can further express the conduction current through as the surface integral
where is the current density vector and
is an infinitesimal area vector normal to the surface.
Differentiating the electric flux through the surface () with respect to time yields
where the time derivative may be moved inside the integral because the surface is assumed to be stationary.
Substituting eq32 and eq33 into eq31 results in:
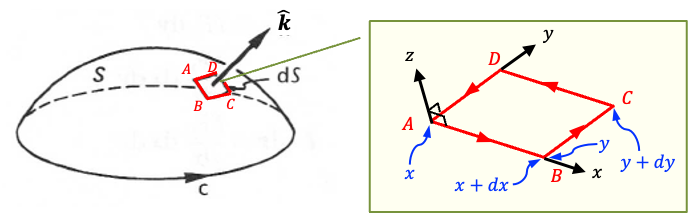
Applying Stokes’ theorem gives:
Since this relation must hold for any arbitrary surface , the integrands must be equal at every point in space. Therefore,
which is the differential form of Maxwell’s fourth equation.
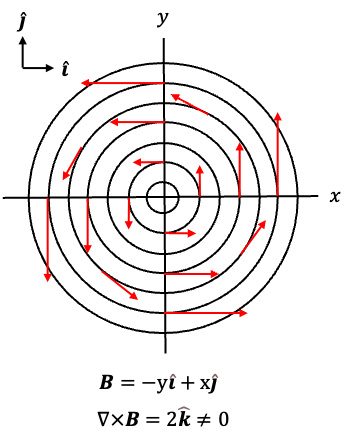
In a vacuum (free space), and eq34 becomes
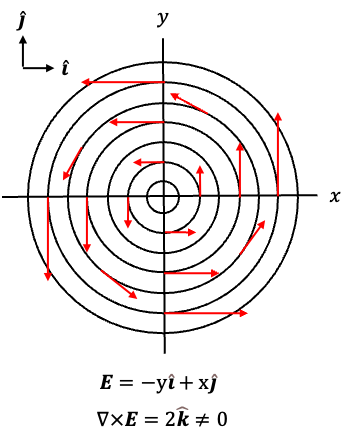
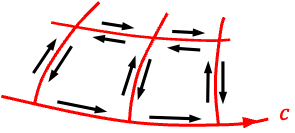
Circulating magnetic field lines are often illustrated using the vector function , or equivalently
(see diagram above), where the field vectors are tangent to circles around the origin and point anticlockwise. For example,
points upwards at
,
points left at
,
points downwards at
, and
points right at
. Since the curl of
is given by
it is non-zero and points in the positive -direction. By the right-hand rule, this corresponds to positive (anticlockwise) circulation of the field in the
-plane. Therefore, eq35 states that a time-varying electric field produces a circulating magnetic field
.
Notably, the curl of is itself a vector:
. If the electric field oscillates with time, then its time derivative
oscillates as well. According to eq35, the curl of the magnetic field
then oscillates with time, causing the vector
to oscillate. The magnetic field
must, in turn, vary with time so that its curl is equal to this oscillating vector. This behaviour is commonly illustrated using an electromagnetic wave diagram, in which electric and magnetic fields vary sinusoidally with time (see diagram below). Although such a diagram may give the impression that the wave is propagating in one direction, it actually shows the temporal oscillation of the electric and magnetic fields at a single point in space. The existence of a propagating electromagnetic wave additionally requires the fields to vary with both position and time.
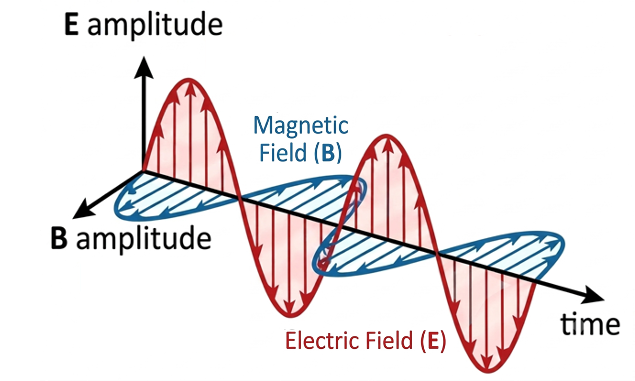
To properly explain electromagnetic wave propagation, we must also consider Maxwell’s third equation . This equation states that a time-varying magnetic field produces a circulating electric field, which is also time-varying. Together with eq35, these two equations describe how a changing electric field generates a changing magnetic field, which in turn generates a changing electric field, and so on This self-sustaining process enables electromagnetic waves to propagate through free space (see diagram below).
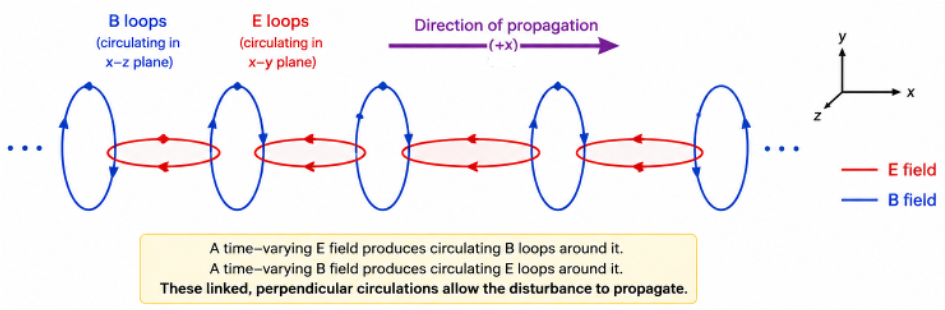
Propagation can also be illustrated using the earlier amplitude diagram, with the time axis replaced by the position () axis.