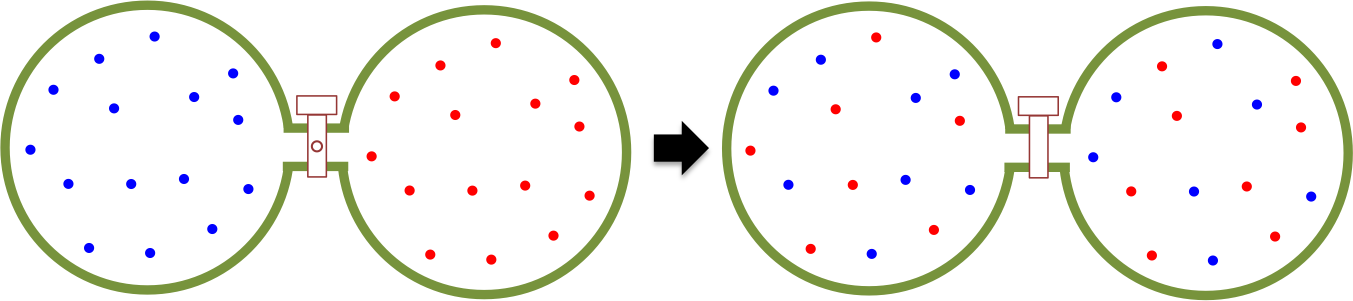
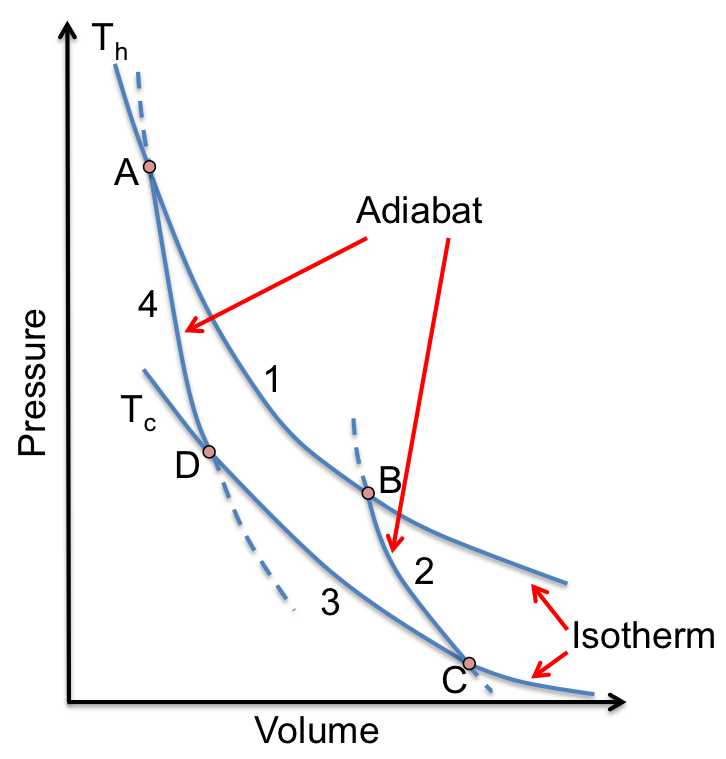
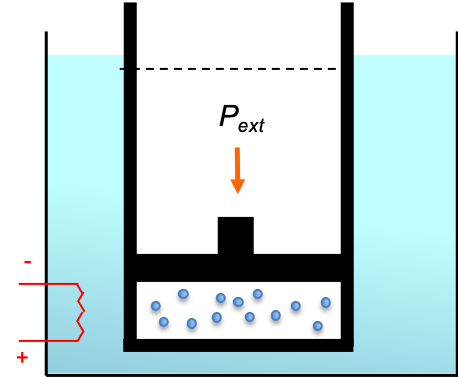
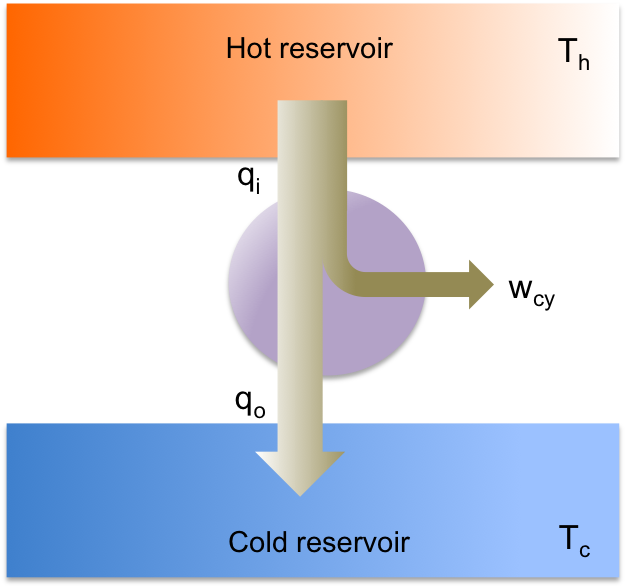
A line integral is the integral of a multi-variable function along a curve. Line integrals can be categorised into scalar line integrals and vector line integrals. They are usually evaluated using parametric equations, which are then reduced to one or more scalar integrals.
Scalar line integrals

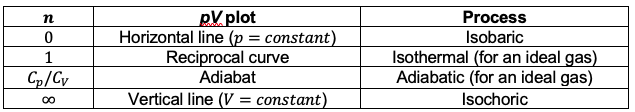
Let’s begin with scalar line integrals. A scalar function of two variables 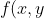) is represented geometrically by a surface on a three-dimensional graph (see diagram above). Consider two points, A and B, on the surface. There are infinite ways to move along the surface between these two points. For simplicity, we have indicated two of these infinite paths using the red and pink curves, which can be projected onto the xy plane for better visualisation (see diagram below).
is represented geometrically by a surface on a three-dimensional graph (see diagram above). Consider two points, A and B, on the surface. There are infinite ways to move along the surface between these two points. For simplicity, we have indicated two of these infinite paths using the red and pink curves, which can be projected onto the xy plane for better visualisation (see diagram below).

Due to the many paths available between the two points, the integral of between the points is not well defined. In other words, the integral is path-dependent. For a particular path, the integral of along this path, which is specified by one of the curves on the 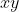 plane, is called a line integral.
plane, is called a line integral.

Geometrically, the result is the area of the ‘curtain’ extending from that path, between ) and
and ) on the xy plane, to the curve on the surface (see diagram above), i.e.:
on the xy plane, to the curve on the surface (see diagram above), i.e.:

where  denotes ‘curve’, which is the specified path linking the two points on the xy plane, and
denotes ‘curve’, which is the specified path linking the two points on the xy plane, and 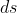 is the infinitesimal change in arc length along the curve.
is the infinitesimal change in arc length along the curve.
Since the specific path is also a function of 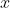 and
and 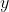 , we convert and into parametric equations consisting of a single parameter and carry out the integration analytically (see 3rd Q&A below for an example).
, we convert and into parametric equations consisting of a single parameter and carry out the integration analytically (see 3rd Q&A below for an example).

Question
What if the two points, A and B, are the same?
Answer
In the scenario where the two points are the same, the infinite paths from to are infinite loops. Similarly, integrating the function via different loops may give different results. Each line integral in this case is represented by:

where the circle on the integral sign denotes a loop or a cyclic process.
Vector line integrals

Let’s now look at vector line integrals, an example of which is the work done by a vector field (see diagram above). For instance, the work done by a variable force  on a charged particle moving along some path in an electric field is
on a charged particle moving along some path in an electric field is
)
As mentioned in the opening paragraph, such a line integral is evaluated by converting it into one or more scalar integrals. We write and  in terms of their two-dimensional Cartesian components:
in terms of their two-dimensional Cartesian components:
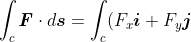\cdot( dx\boldsymbol{i}+dy\boldsymbol{j})=\int_cF_xdx+\int_cF_ydy)
which can then be evaluated when is specified.
Question
What is the difference between a scalar field and a vector field?
Answer
A scalar field, expressed by a scalar function , associates a scalar with each point in some region of space, while a vector field, expressed by a vector function ) associates a vector with each point.
associates a vector with each point.
Line integral with respect to coordinates

Line integrals can also be carried out with respect to one of the function variables instead of with respect to the arc length, e.g. the scalar integral  . The geometric interpretation of is the projection of on the
. The geometric interpretation of is the projection of on the  plane (see above diagram). An example of such a line integral is the work done on an ideal gas in a reversible process:
plane (see above diagram). An example of such a line integral is the work done on an ideal gas in a reversible process:
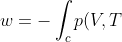dV)
If the path of the above integral is defined by introducing a constraint, e.g. when 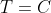 , where
, where 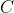 is a constant, we get a plane that intersects with the surface at a particular contour (see diagram below). A single curve, an isothermal curve, is then projected on the
is a constant, we get a plane that intersects with the surface at a particular contour (see diagram below). A single curve, an isothermal curve, is then projected on the  plane.
plane.

Consequently, the integral reduces to one involving a single variable:
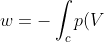dV=-nRT\int_{V_1}^{V_2}\frac{1}{V}dV)
Similarly, if the intersecting plane is  or
or 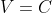 , the projection on the plane is a horizontal line (isobaric process) or a vertical line (isochoric process) respectively.
, the projection on the plane is a horizontal line (isobaric process) or a vertical line (isochoric process) respectively.
Line integral of differential forms
Line integrals are sometimes written in differential form. Consider the work done by a vector field on a particle moving along some path ) . We can write and
. We can write and  in terms of their components:
in terms of their components:  and
and  . So, eq92 becomes
. So, eq92 becomes
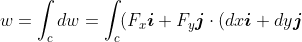=\int_c\left(F_xdx+F_ydy\right))
Compared to the general differential equation of two independent variables of the form dx+N(x,y)dy) , the RHS of the third equality of the above equation is the line integral of a differential equation, which in this case is an inexact differential. In other words,
, the RHS of the third equality of the above equation is the line integral of a differential equation, which in this case is an inexact differential. In other words, =\int_cdu) .
.
In the case of an exact differential  , its line integral is equal to the difference in values of the function
, its line integral is equal to the difference in values of the function 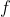 at the final point and at the starting point:
at the final point and at the starting point:
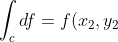-f(x_1,y_1)=\Delta f\;\;\;\;\;\;\;\;(93))
This is known as the fundamental theorem of line integral. We call such a function, whose output is independent of the path taken to reach it, a state function.
Finally, we have shown in a previous article that the change in internal energy of a system  is path-independent for a system containing a perfect gas. We will show in the article on entropy that is path-independent for any system. This makes
is path-independent for a system containing a perfect gas. We will show in the article on entropy that is path-independent for any system. This makes  an exact differential and
an exact differential and 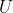 , a state function. Thus, we can write
, a state function. Thus, we can write
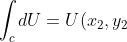-U(x_1,y_1)=\Delta U)
If the two points are the same, =(x_1,y_1)) ,
,
)
Therefore, the change in internal energy in a cyclic process is zero:  . In general, the line integral of any exact differential involved in a cyclic process is zero.
. In general, the line integral of any exact differential involved in a cyclic process is zero.