Johannes van der Waals, a Dutch physicist, introduced an equation in 1873 called the van der Waals equation to mathematically describe the physical state of a real gas.
This equation is:
![\left [ p+a\left ( \frac{n}{V} \right )^2 \right ]\left ( V-nb \right )=nRT\; \; \; \; \; \; \; \; 3](https://latex.codecogs.com/gif.latex?\left&space;[&space;p+a\left&space;(&space;\frac{n}{V}&space;\right&space;)^2&space;\right&space;]\left&space;(&space;V-nb&space;\right&space;)=nRT\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;3)
where a and b are called the van der Waals coefficients, which are temperature-independent constants specific to each gas.
The equation looks similar to the ideal gas equation except for the pressure and volume factors of ^2) and nb, respectively. To understand how van der Waals’ equation is derived, we need to know how the ideal gas law is conceptualised.
and nb, respectively. To understand how van der Waals’ equation is derived, we need to know how the ideal gas law is conceptualised.
The ideal gas equation, pV = nRT, is derived from the combined experiments of Robert Boyle and Jacques Charles, and a principle developed by Amedeo Avogadro. This means that, for a fixed amount of gas at a relatively high temperature, the product of the experimentally observed values of p and V in the equation is a constant. The equation works reasonably well at low pressures where gases approach ideality. Under such conditions, gas particles can be assumed to be point masses with zero volume and, therefore, have no intermolecular forces of attraction or repulsion between them. In other words, the equation only works when the observed variables of p and V are ‘ideal’:

In reality, gases have finite volumes and repel one another, especially at high pressures. The experimentally observed values of pressures and volumes are actually preal and Vreal. Therefore, for eq4 to work, we have to modify it.
Due to the presence of repulsive forces between real gas molecules at high pressures, the observed volume of a real gas is larger than the volume of its ideal counterpart, that is, Vreal > Videal. The diagram below shows the volume expansion (from yellow to blue) if the point masses have finite volumes.
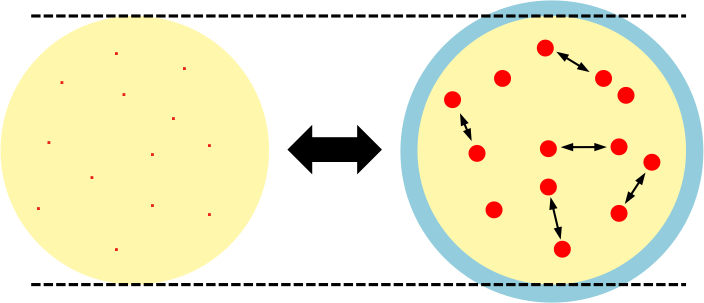
Letting the difference in molar volume of a real gas and an ideal gas be b, we have  or
or

Substituting eq5 in eq4 and rearranging yields

Eq6 predicts that the molar volume of a gas at zero Kelvin is b. Experiments indeed reveal that b has approximately the same value as the molar volume of the solid form or liquid form of the gas near that temperature. On the other hand, the ideal gas law,  , wrongly predicts that the volume of a gas at 0 K is zero.
, wrongly predicts that the volume of a gas at 0 K is zero.
Next, the pressure that a real gas exerts on the wall of a container is lower than the pressure of its ideal counterpart (pideal > preal), as the molecules of a real gas experience intermolecular forces of attraction that decelerates their motion when they are about to impact the wall of the container. This means that we need to add a pressure shortfall, Δpreal, to the observed real pressure to reflect the ideal pressure that fits eq4:

If we perceive the intermolecular force of attraction (i.e., the force that one molecule of gas experiences near the wall of the container) as the attraction between that molecule and the bulk of the gas, the pressure shortfall due to that particular molecule is proportional to the density of the gas:
)
Furthermore, the total number of molecules N near the wall of the container is again proportional to the density of the gas:
)
The total pressure shortfall is therefore:
k\left&space;(&space;\frac{n}{V_{real}}&space;\right&space;)=a\left&space;(&space;\frac{n}{V_{real}}&space;\right&space;)^2)
where a = k’k is a proportionality constant specific to a particular gas.
Eq7 becomes
^2\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;8)
Substituting eq5 and eq8 in eq4, we get the explicit form of eq3:
![\left [ p_{real}+a\left ( \frac{n}{V_{real}} \right )^2 \right ]\left ( V_{real}-nb \right )=nRT\; \; \; \; \; \; \; \; 9](https://latex.codecogs.com/gif.latex?\left&space;[&space;p_{real}+a\left&space;(&space;\frac{n}{V_{real}}&space;\right&space;)^2&space;\right&space;]\left&space;(&space;V_{real}-nb&space;\right&space;)=nRT\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;9)
The pressure and volumes in eq9 now represent experimentally observed real pressure and real volumes, respectively. We have essentially converted an equation that only works for ideal gases into one that is applicable for real gases. For simplicity, we can omit the subscripts, which gives eq3.
Using eq3, the van der Waals equation can also be expressed in the molar form by substituting it with the molar volume,  , to give:
, to give:
\left&space;(&space;V_m-b&space;\right&space;)=RT)
which rearranges to:

At high molar volumes (i.e. low pressures), Vm – b ≈ Vm and the second term on the RHS of eq10 becomes very small, with eq10 reducing to the ideal gas equation:  .
.
The van der Waals equation is an improvement over eq2, as it is able to describe the physical state of a real gas in terms of the van der Waals coefficients a and b, which relate to the strength of the forces of attraction between molecules and the size of the molecules, respectively.
Since the critical isotherm of a gas has an inflexion point at the critical point, we can determine the van der Waals coefficients for a gas by finding the 1st and 2nd derivatives of eq10, letting these be equal to zero and solving for the critical constants:
^2}+\frac{2a}{V_m^{\:&space;3}}=0\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;11)
^3}-\frac{6a}{V_m^{\:4}}=0\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;\;&space;12)
From eq11 and eq12, ^2}) and
and ^3}) respectively. Combining both values of a,
respectively. Combining both values of a,

Substitute eq13 back in eq11 where Vm is now Vc,

Substitute eq13 and eq14 back in eq10,

Hence, by measuring the pressure, volume and temperature at the critical point of a gas (i.e. the critical constants), we can calculate the van der Waals coefficients for that gas. The table below shows the van der Waals coefficients for some common gases:
|
|
a / atm dm6 mol-2 |
b / 10-2 dm3 mol-1
|
|
H2
|
0.2420 |
2.65 |
|
N2
|
1.352 |
3.87
|
|
O2
|
1.364 |
3.19
|
| CO2 |
3.610 |
4.29
|