Mass–energy equivalence is the principle that mass can be converted into energy and energy into mass, with the relationship quantified by E = mc2.
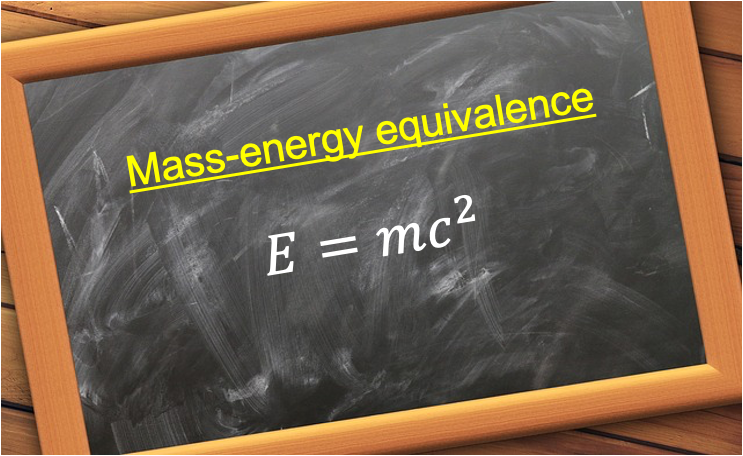
The idea of mass–energy equivalence emerged from attempts to resolve inconsistencies between classical physics and electromagnetism in the early 20th century. Before Einstein, physics treated mass and energy as completely separate quantities: mass was seen as “matter content,” while energy was something objects had due to motion, heat or fields. However, developments in electromagnetism (especially Maxwell’s equations) and experiments with light showed that energy itself carries momentum, hinting that energy and mass might be more deeply connected.
In 1905, Albert Einstein published his special relativity paper and later that year derived a key consequence: if an object emits energy (like light), its mass must decrease by an amount proportional to the energy released divided by the speed of light squared. This led to the compact relation , where
is the speed of light. Later experiments in nuclear physics confirmed the idea dramatically: in processes like nuclear fission and fusion, small losses of mass are converted into enormous amounts of energy, matching Einstein’s equation with high precision.
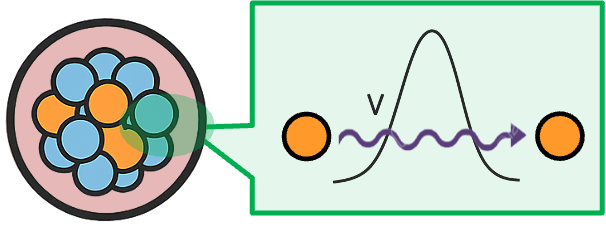
To derive the relation, we refer to Einstein’s thought experiment, which uses the principles of conservation of momentum and conservation of the centre of mass. Consider a hollow, completely closed box of mass and length
, floating at rest in deep, frictionless space. Suppose a photon of energy
is spontaneously emitted from the left wall towards the right wall (see diagram below). The momentum of the photon is given by
, which can be derived by substituting
and de Broglie’s relation
into Planck’s law
.
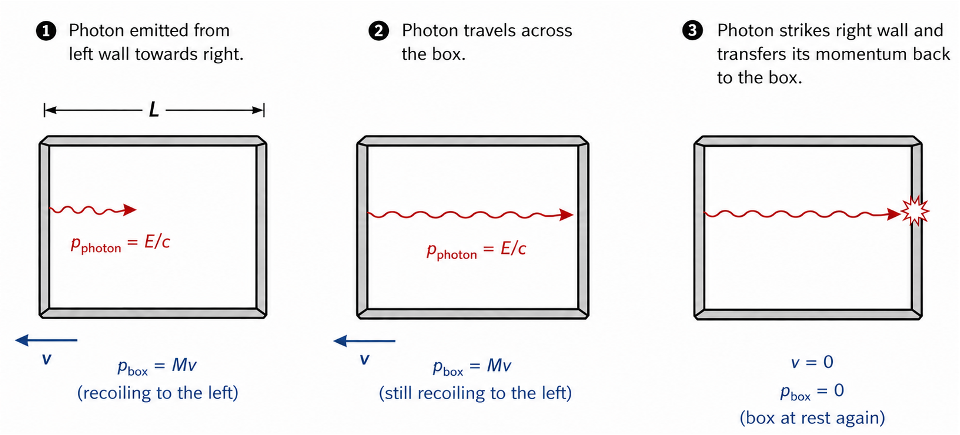
According the law of conservation of momentum, the emission of the photon causes the box to recoil to the left with velocity and momentum
. It follows that
, or equivalently,
As the photon travels across the box and strikes the right wall, it transfers its momentum back to the box, bringing the box to rest again. Thus, the box starts at rest and ends at rest, but during the photon’s transit it shifts leftwards by a very small distance , where
is the time taken for the photon to travel across the box. Hence,
Substituting eq20 and eq21 into gives:
Since no external force acts on the system, the centre of mass must remain stationary. To resolve the apparent contradiction caused by the box’s displacement, Einstein concluded that the energy carried by the photon must possess an inertial equivalent mass from the left side of the box to the right side. The shift in the photon’s equivalent mass balances the displacement of the box:
Substituting eq22 into eq23 and rearranging yields the Einstein relation:
This result is not limited to photons, since the photon merely serves as a convenient carrier of energy across the box. Einstein’s thought experiment considers the box-photon system as a whole in its rest frame. Before the photon is emitted and after it is absorbed, the total system remains stationary. While the photon itself moves within the box, its energy remains part of the total energy of the closed system. The moving photon therefore contributes to the system’s total rest mass according to . In other words, the equation applies to the total energy contained in a system that is overall at rest, even if energy is moving internally within that system. It demonstrates a more general principle: any form of energy possesses inertia and therefore contributes to the rest mass of a system. For example, a heated object possesses slightly greater mass than the same object when colder. In this case,
refers to the total rest mass of the entire object as a system, including both the masses of its constituent particles and their internal energy. The hotter object contains additional thermal energy associated with the motion and interactions of its particles, so its total mass is slightly greater. The energy difference between the hotter and cooler object is therefore given by
.

Question
Does the speed of light in contradict the fact that the formula applies to the total energy of a system in its rest frame?
Answer
No. Here, is a fundamental constant, and
acts as a conversion factor between mass and energy. It is similar to how
does not mean that an object must be falling with acceleration
. For example, a stationary battery has slightly greater mass when it is fully charged than when it is empty, because stored energy contributes to the battery’s mass even though the battery is not moving at all.