Vibronic and ro-vibronic selection rules specify which combined electronic, vibrational, and rotational transitions in molecules are allowed based on quantum number and symmetry constraints.
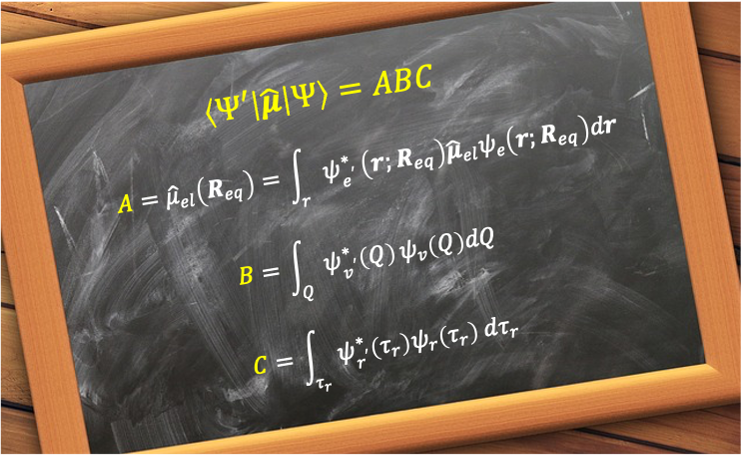
An electronic transition in a molecule is often accompanied by vibration-rotation transitions. To determine the selection rules for these transitions, we refer to the Born-Oppenheimer approximation, which states that the total wavefunction of the molecule can be expressed as:
where
is the electronic wavefunction that is a function of nuclear coordinates
and electronic coordinates
.
is the vibrational wavefunction that is a function of normal coordinates
.
is the rotational wavefunction that is a function of rotational coordinates
, typically the Euler angles.
According to the time-dependent perturbation theory, the transition probability between the initial and final states, and
is proportional to the square of
, where the electric dipole moment operator
is the sum of all charge–position contributions in the molecule:
where
and
are the charge and position of nucleus
.
and
are the charge and position of electron
.
The total transition matrix is:
Although depends parametrically on the nuclear coordinates, the nuclear dipole operator
does not act on the electronic coordinates. Therefore, we can write:
Since electronic spectroscopy involves transitions between different electronic states, and the eigenfunctions in the set are orthonormal,
and thus this term vanishes. In contrast, for microwave or infrared spectroscopy, where no change in electronic state occurs,
, and so:
which corresponds to vibration-rotation transitions.
Therefore, the total transition matrix reduces to:
Because the integral in square brackets is performed over all electronic coordinates for each set of nuclear coordinates , we can define it as a function of
:
. To further simplify the total transition matrix, we apply the Condon approximation, which assumes that electronic transitions occur so rapidly (on the order of 10-15 s) that the nuclei can be considered stationary during the transition. Evaluating at the equilibrium nuclear geometry
(i.e. an average internuclear separation),
can be treated as a constant. Thus,

Question
Is defined in the molecular frame or the laboratory frame?
Answer
It is defined in the molecular frame. In the Born-Oppenheimer approximation, the electronic coordinates are defined relative to the nuclear centre of mass.
Although is defined in the molecular frame
, the rotational wavefunctions are defined with respect to the laboratory frame
. For the integral to be physically meaningful, the dipole operator and the wavefunctions must be expressed in the same coordinate system (usually the laboratory frame) as that is where the interaction with the external electromagnetic field occurs. To simplify the integral, we shall consider the projection of
onto the lab
-axis (see this article for derivation):
where ,
,
.
Since ,
,
are constants (Condon approximation),
with (see this article for derivation).
defines the electronic selection rules. As explained in the previous article, these rules are governed by
-
- Symmetry: One first identifies the point group of the molecule, then determines the symmetries of the initial and final states, and finally applies the vanishing integral rules.
- Parity: The Laporte selection rule applies only to molecules possessing an inversion centre (e.g. those belonging to
or
).
- Spin:
, assuming spin-orbit coupling is weak.
For example (see previous article for derivation),

Unlike pure molecular vibrational motion in infrared spectroscopy, where the initial and final states are part of a complete set of eigenfunctions of the same Hamiltonian (resulting in the pure vibrational selection rule ), the initial state in
(known as the Frank-Condon overlap integral) belongs to a complete set of eigenfunctions of a Hamiltonian that is different from that of the final state. This is because the two states correspond to different electronic states, each with a different potential-energy function. Therefore, the final vibrational state may not be orthogonal to the initial vibrational state. However, both complete sets of eigenfunctions span the same Hilbert space, which implies that we can expand
as a linear combination of
:
with
If there were a restriction such as , the expansion
would contain only a finite number of terms. However, the excited state generally requires infinitely many initial state basis functions to represent. This corresponds to an infinite number of nonzero coefficients
and hence infinitely many possible values of
, resulting in no selection rule for
. In other words,
For linear molecules, and
, while
, even for homonuclear diatomic molecules, because of the generation of a transient dipole moment in electronic spectroscopy. Therefore,
in
simplifies to
, where the spherical harmonics are the wavefunctions, and the volume element reduces to
because the spherical harmonics do not depend on
. As shown in another article, this integral yields the selection rules:
. The inclusion of
reflects the fact that conservation of total angular momentum is satisfied during a photon-mediated electronic transition in linear molecules when
, even though
is forbidden for pure rotational transitions in linear molecules. More specifically, the ro-vibronic transition selection rules for linear molecules are:
This is why homonuclear diatomic molecules are vibrationally and rotationally inactive in standard infrared and microwave spectroscopy, but vibrationally and rotationally active in electronic spectroscopy.
For symmetric rotors, the rotational wavefunctions are represented by the Wigner D-functions . As shown in another article,
results in
for parallel transitions, and
for perpendicular transitions. Therefore, for allowed electronic transitions,
Although a spherical top is rotationally inactive in pure rotational spectroscopy, it is rotationally active in ro-vibronic transitions due to the generation of a transcient dipole moment. Since a spherical top is a special case of a symmetric top for which the energy does not depend on , the selection rules for allowed electronic transitions are simply: