The mesomeric effect is the electron-donating or electron-withdrawing effect of a substituent transmitted through a conjugated π-system by resonance.
Resonance, or the resonance effect, typically occurs when atoms are connected by alternating single and multiple bonds, where electrons can be shared across several atoms instead of being localised. For instance, the π-electrons in benzene are dislocalised over the entire ring rather than confined to three double bonds depicted in a Lewis structure.
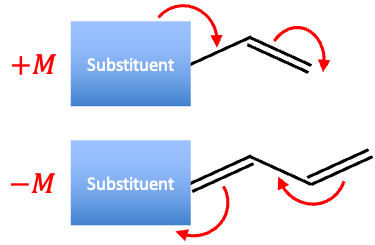
In other words, the mesomeric effect is an electron-donating (+M) or electron-withdrawing (-M) effect via resonance. It requires a functional group attached to a conjugated system and focuses on how the group changes the electron distribution.

Question
Is the mesomeric effect different from the inductive effect?
Answer
Yes, the inductive effect is the displacement of electron density through -bonds along a chain of atoms due to the presence of an electronegative or electropositive atom or group. In contrast, the mesomeric effect involves the delocalisation of electrons through π-bonds. Although the mesomeric effect is governed mainly by resonance and orbital interactions, the electronegativities of atoms in substituents may be considered when analysing the mesomeric effect of a molecule (e.g. via the Hückel method). Nevertheless, the two effects are generally different. For example, lone pairs on atoms such as O or N often give rise to a +M effect. However, these same atoms may exert a –I effect at the same time. Therefore, the mesomeric and inductive effects can act in the same direction or in opposite directions in a molecule with a substituent.
-M effect
The Hückel method is the standard semi-empirical approach to quantify the mesomeric effect in a conjugated system. Consider the acrolein molecule CH2=CH-CHO, which contains two π-bonds, one of which belongs to an electron-withdrawing formyl group (-CHO). In general, we can express the one-electron molecular orbital (MO) wavefunction of acrolein as:
where and
represent the coefficients and the p atomic orbital wavefunctions respectively;
corresponds to the oxygen atom,
corresponds to the carbonyl carbon,
corresponds to the middle ethylene carbon and
corresponds to the terminal ethylene carbon.
One quick way to analyse the MO energies of acrolein is to refer to the secular determinant of butadiene:
which represents a four- atom framework with two alternating π-bonds.
To account for the electronegativity of oxygen, we modify the Hückel parameters as follows:
where
is the Coulomb integral for oxygen, with
being a factor that lowers the orbital energy.
is the Coulomb integral for the carbonyl carbon.
is the resonance integral for overlap between C and O.
is the Coulomb integral for carbon atoms.
is the resonance integral between carbon atoms.
is the energy of an MO.
The next step involves estimating trial values for ,
and
. Since oxygen is more electronegative than carbon, we set
. This results in C=O being a stronger bond compared to C-C. Consequently, we set
. The corresponding inductive effect by oxygen on the the carbonyl carbon causes it to be slightly electronegative relative to the adjacent ethylene carbon. We can therefore set
. It is important to note that although the electronegativity of oxygen helps stabilise the system, it is not the primary cause of the mesomeric effect, which is fundamentally due to conjugation and orbital overlap.
Using the matrix identity of , and multiplying both sides of eq390 by
, give
where .
Expanding the determinant gives the characteristic equation:
with solutions , which when substituted back into
yields:
| MO | Type | ||
| 1.52 | Antibonding | ||
| 0.31 | LUMO | ||
| -1.16 | HOMO | ||
| -2.86 | Bonding |
Question
Show that the isolated C=O Hückel bond energy is .
Answer
The secular determinant for the C=O bond is
Solving for gives
. Therefore, the bonding MO energy is
.
The total ground state π-electron energy, , is lower than the sum of the energies of isolated C=C (
) and C=O (
) bonds by
, indicating the delocalisation energy provided by the mesomeric effect. To further understand the electron distribution of the molecule, we determine the coefficients of the
wavefunctions as follows:
which implies:
The simultaneous equations for can be solved by substituting
into them and expressing all coefficients in terms of
:
Normalisation gives:
Therefore, . Repeating the calculations for
and
yields:
| Coefficient | Ground state electron density | |||
| c4 | 0.07 | 0.60 | 0.67 | 0.73 |
| c3 | 0.20 | 0.70 | -0.21 | 1.06 |
| c2 | 0.50 | 0.21 | -0.60 | 0.59 |
| c1 | 0.82 | -0.35 | 0.37 | 1.59 |
where the ground state electron density is given by for two electrons in each MO.
The value of 1.59 at oxygen indicates significant accumulation of π-electron density. This arises from the –M effect of the carbonyl group, where electron density is delocalised towards the oxygen atom. In contrast, the terminal carbon and the carbonyl carbon are electron deficient due to this delocalisation (see resonance structures below). While oxygen’s electronegativity contributes to the overall polarisation of the molecule, the calculated electron densities primarily reflect the mesomeric effect rather than the inductive effect. In this case, both effects act in the same direction.
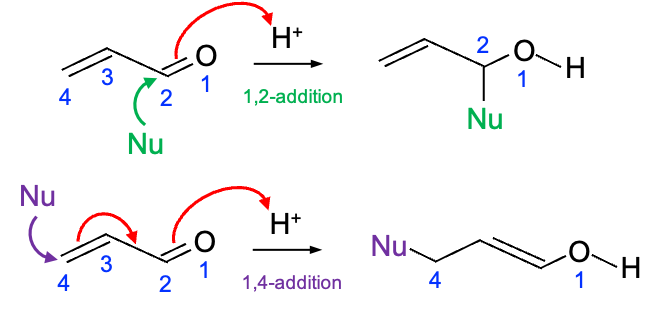
The calculations also suggest regioselectivity of acrolein in reactions with nucleophiles. For example, “hard” (ionic) nucleophiles, such as LiAlH4 or Grignard reagents, are driven mainly by electrostatic attraction and attack the most electron-deficient carbonyl carbon (with ground state electron density of 0.59), resulting in 1,2-addition reactions (see diagram below). On the other hand, “soft” (covalent) nucleophiles, like enolates, thiols or organocuprates, are governed by orbital interactions. They attack the atom where the LUMO has the largest “lobe” (), which is the terminal ethylene carbon of acrolein, leading to 1,4-addition (Michael addition) reactions.
+M effect
The +M effect is typically attributed to a substituent with lone pair electrons. Consider vinylamine CH2=CH-NH2, with the general one-electron MO wavefunction:
where represents the p atomic orbital wavefunctions; 1 denotes the nitrogen atom, 2 denotes the middle ethylene carbon and 3 denotes the terminal ethylene carbon.
The secular determinant is given by:
where
is the Coulomb integral for nitrogen, with
being a factor that lowers the orbital energy.
is the resonance integral for overlap between C and N.
is the Coulomb integral for carbon atoms.
is the resonance integral between carbon atoms.
is the energy of an MO.
Let’s set due to the electronegativity of N, and
because the C-N bond is slightly weaker than the C=C bond. This translates to:
with the corresponding characteristic equation:
Substituting the solutions back into
yields:
| MO | Type | ||
| 1.13 | LUMO | ||
| -0.68 | HOMO | ||
| -1.95 | Bonding |
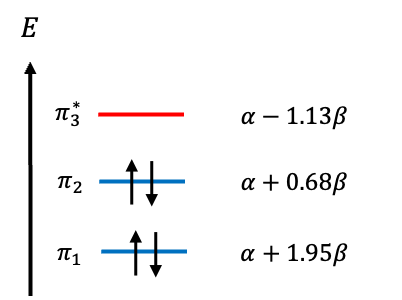
Unlike acrolein, in which one electron from each of the four atoms populates the MOs, vinylamine has two nitrogen electrons (lone pair) and one electron from each carbon atom contributing to its bonding and highest occupied MOs. The total ground state π-electron energy, , is lower than the sum of the energies of isolated C=C bond (
) and the nitrogen lone-pair (
), indicating a delocalisation energy of
provided by the +M effect.
Question
Show that the isolated nitrogen lone pair energy is .
Answer
Since nitrogen’s lone pair is not yet delocalised into the π-system, its energy corresponds to twice the Coulomb integral , which is
.
The coefficients of the wavefunctions are given by:
Substituting the three values of into the matrix equation and solving it gives:
| Coefficient | Ground state electron density | |||
| c3 | 0.24 | 0.72 | -0.65 | 1.15 |
| c2 | 0.48 | 0.49 | 0.73 | 0.94 |
| c1 | 0.85 | -0.48 | -0.22 | 1.91 |
Since the ground state electron density of a neutral, non-polarised one-electron atom is 1.0, the ground state electron density of the nitrogen lone pair of 1.91 is lower than its unpolarised value of 2.0 (density = number of electrons per unit volume). In contrast, the electron density of the terminal carbon of 1.15 is higher than its non-polarised value of 1.0. This shift of electron density from N to the terminal C is a consequence of the +M effect of the amine group, even though N exerts a –I effect on its neighbours. In this case, mesomeric and inductive effects act in opposite directions. Furthermore, the relatively electron-rich terminal carbon and nitrogen atoms (ground state electron density > 1.0) make the molecule susceptible to electrophilic attack.
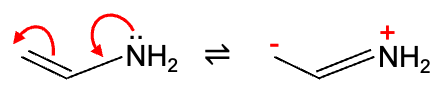
In terms of regioselectivity, a “hard” electrophile like H+ (small, concentrated positive charge) is attracted to N, which has the highest ground state electron density of 1.91. Protonation at N forms an ammonium ion. H+ can also attack the terminal carbon (electron density = 1.15) to yield an iminium ion. The relative proportion of ammonium and iminium ions formed depends on temperature and reaction time.
The ammonium ion forms quickly at relatively low temperatures through fast collisions and is considered the kinetic product. However, it is not energetically favourable because formation of the -bond between H+ and N disrupts the conjugated system, reducing the molecule’s delocalisation energy. Given enough time for equilibration at room temperature, the resonance-stabilised iminium ion, in which the positive charge is delocalised between carbon and nitrogen, becomes the thermodynamic product and predominates. If water is present, this iminium ion quickly hydrolyses to acetaldehyde and an ammonium salt, which explains why vinylamines (and enamines) are often used as intermediates to functionalise carbonyls.
On the other hand, a “soft” electrophile such as CH3I, in which the the partially positive carbon is diffuse due to iodine’s polarisability, seeks the largest HOMO lobe for orbital overlap. This leads to formation of a new carbon-carbon bond at the terminal carbon of vinylamine.
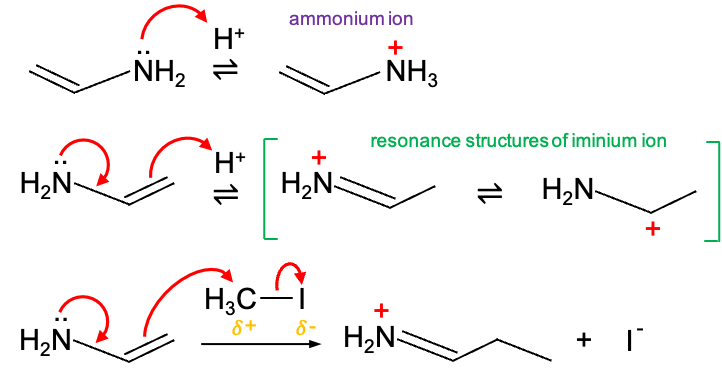
Overall, the reactivity of vinylamine illustrates how electron density, substituent effects and the nature of the electrophile work together to determine both regioselectivity and product distribution. Understanding these effects allows chemists to predict and control reaction outcomes, making vinylamines valuable intermediates in organic synthesis.