Infrared (IR) spectroscopy measures the interaction of infrared radiation with vibrating molecules, allowing the identification of chemical substances or functional groups.
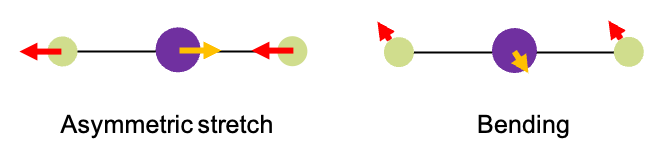
Molecules consists of atoms connected by chemical bonds. These bonds can vibrate – stretching, bending and twisting – when the molecules absorb specific wavelengths of IR radiation. For instance, when absorbs a specific frequency of IR radiation, it undergoes a vibrational mode known as an asymmetric stretch (see diagram above). During this motion, the two oxygen atoms vibrate periodically in a direction opposite to that of the carbon atom. However, If it absorbs a different IR frequency, the carbon and oxygen atoms move perpendicularly in and out of the screen. Notably, the carbon atom moves in opposite direction to the oxygen atoms. We refer to this type of vibrational mode as bending.

Question
Why does a molecule vibrate when it absorbs radiation at the IR range and not at other frequencies of the electromagnetic spectrum?
Answer
When atoms within a molecule vibrate, the electron distribution in the bonds connecting them changes. Similar to the energy needed to compress two balls held by a spring of a specific material, a precise amount of energy is required to redistribute the electron density of two bonded atoms. This energy’s frequency falls within the IR range of the electromagnetic spectrum.
Each molecule has a unique set of vibrational modes. When IR radiation interacts with a molecule, it selectively excites these vibrational modes. The IR spectrometer measures the absorbance of IR radiation by a molecule and provides an absorption spectrum with peaks at different frequencies. By comparing a sample’s absorption peaks to a database of known spectra, scientists can identify the molecule.
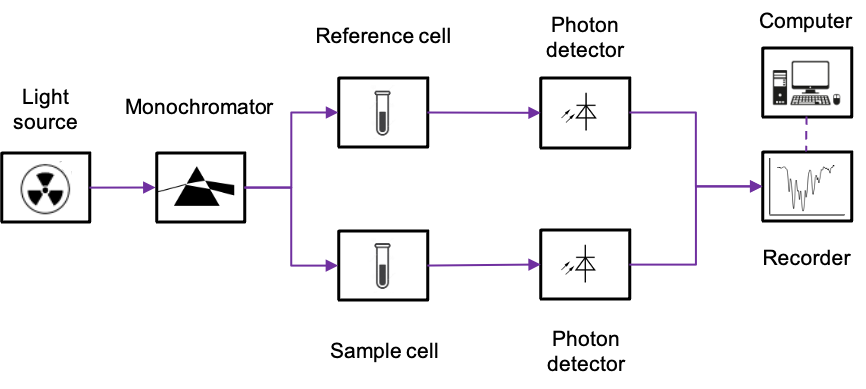
There are two commonly used instruments that measure the interaction of IR radiation with molecules – dispersive IR spectrometer and Fourier-transform IR spectrometer. The diagram below outlines a typical double-beam dispersive IR spectrometer, which utilises a heated rod, wire or coil as the radiation source. The materials used for fabricating these components can be rare-earth oxides (for the Nernst Glower), silicon carbide (for the Globar) or nichrome (for the Nichrome Coil). All of these materials produce continuous radiations when heated to specific temperatures within the range of 1000 oC to 1800oC.
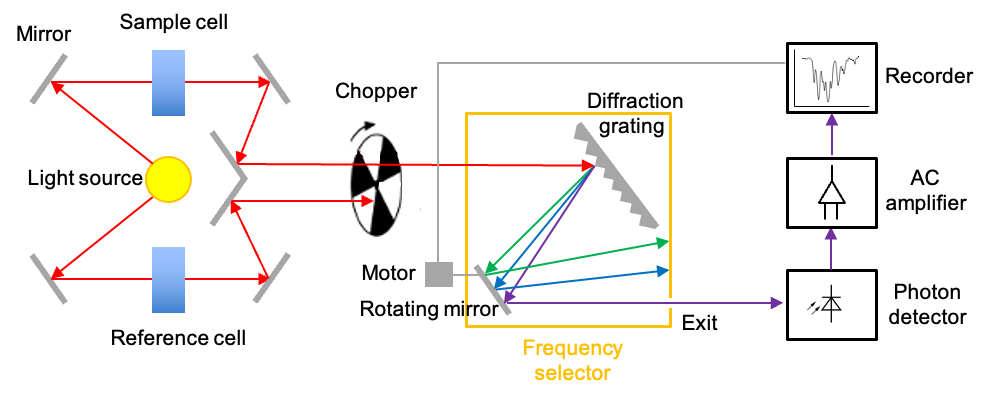
A beam of broad spectrum infrared light produced is first split into two separate beams by mirrors. One beam passes through the sample cell, and the other through a reference cell. For an aqueous sample, the reference cell is filled with the corresponding pure solvent; otherwise it is empty. A rotating chopper periodically blocks one of the beams, allowing the other to reach the diffraction grating, which disperses the radiation into its component frequencies. With the help of a rotating mirror, the component frequencies of the dispersed spectrum are then scanned across the exit, beyond which lies the photon detector.
The photodetector operates based on the photoelectric effect. This is where the interaction of IR radiation with a semiconductor promotes electrons to the conduction band, generating a small current that is proportional to the intensity of the radiation. When the sample absorbs at a specific component frequency, the reference and sample signals reach the detector periodically. Since the detector generates a larger current every time it receives a reference signal and a smaller current when it receives a sample signal, it produces an alternating current (AC) that has the same frequency as the rotating chopper.
The AC signal is proportional to the absorption intensity at a specific component frequency. If the sample does not absorb at that frequency, the intensities of the sample and reference beams reaching the detector are equal. In this case, the AC operational amplifier, which is designed to pass only AC signals, provides no output. Finally, the motor that rotates the mirror also moves the chart paper. This movement allows the spectrum to be plotted as a function of frequency or wavenumber.
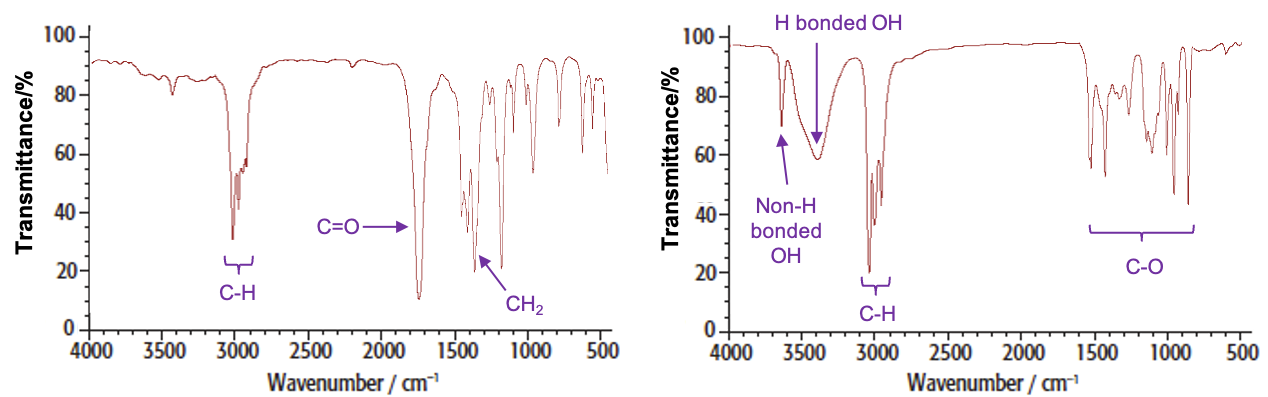
The dispersive IR spectrometer is particularly useful for identifying functional groups of organic compounds. Some characteristic absorption ranges are as follows:
|
Bond |
Functional group |
Absorption wavenumber/ cm-1 |
Appearance of peak |
|
Alcohols, ethers, esters |
1040-1300 | Strong | |
|
Aromatic compounds, alkenes |
1500-1680 | Weak unless conjugated | |
|
Amides Ketones & aldehydes Esters |
1640-1690 1670-1740 1715-1750 |
Strong Strong Strong |
|
|
Alkynes |
2150-2250 |
Weak unless conjugated |
|
| Nitriles | 2200-2250 |
Weak |
|
| Alkanes
Alkenes & arenes |
2850-2950
3000-3100 |
Strong Weak |
|
|
Amines, amides |
3300-3500 | Weak | |
| Carboxylic acids
H-bonded alcohols Non H-bonded alcohols |
2500-3000
3200-3600 3580-3650 |
Strong and broad Strong and broad Strong |
For example, the spectra of butanone and butan-2-ol are: