The solubility product of a chemical compound is the equilibrium constant for the dissolution of the compound in its solid state in an aqueous solution.
The solubility of a solute is the maximum amount of the solute in grammes that can dissolve in a 100 ml (or sometimes 1 dm3 or 1 kg) of a solvent at a particular temperature. For example, the solubility of AgCl in 100 ml of water at 25oC is about 1.92×10-4 g or 1.34×10-5 moles.
To illustrate the concept of solubility product, let’s begin by adding 1.0×10-5 moles of solid AgCl in 100 ml of water at 25oC. The solid completely dissolves, producing 1.0×10-5 moles each of Ag+ and Cl– ions. As we increase the amount of solid AgCl to 1.34×10-5 moles, it still dissolves completely, yielding the maximum concentration of Ag+ and Cl– ions possible in 100ml of water. At this point, the solution is considered saturated. If we add more than 1.34×10-5 moles of AgCl, any excess solid will remain undissolved. The concentrations of Ag+ and Cl– ions stay constant at 1.34×10-5 mol per 100 ml, and the system reaches equilibrium. At equilibrium, the rate at which solid AgCl dissolves into ions equals the rate at which the ions recombine to form the solid, establishing a dynamic balance:
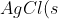\rightleftharpoons&space;Ag^+(aq)+Cl^-(aq))
Since the concentration of solid silver chloride is assumed to be the same as that of its pure state, the equilibrium constant is:
![K_{sp}=[Ag^+][Cl^-]](https://latex.codecogs.com/gif.latex?K_{sp}=[Ag^+][Cl^-])
with
Ksp being the solubility product of AgCl, which is 1.8×10-10 at 25oC.
[Ag+] is the maximum amount of Ag+ in 100 ml of water.
[Cl–] is the maximum amount of Cl– in 100 ml of water.
The solubility product of AgCl is therefore the mathematical product of the solubility of Ag+ (with respect to Cl–) and the solubility of Cl– (with respect to Ag+) raised to the power of their respective stoichiometric coefficients in 100 ml of solvent at a particular temperature. In general, the solubility product of AxBy is
![K_{sp,A_xB_y}=[A^{y+}]^x[B^{x-}]^y](https://latex.codecogs.com/gif.latex?K_{sp,A_xB_y}=[A^{y+}]^x[B^{x-}]^y)

Question
Calculate the Ksp for PbCl2, given that its solubility is 0.0108 g/ml at 20oC.
Answer
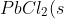\rightleftharpoons&space;Pb^{2+}(aq)+2Cl^-(aq))
Solubility of Pb2+ with respect to Cl– = ![\frac{10.8}{\left [ 207.2+\left ( 2\times 35.45 \right ) \right ]}\; mol\, dm^{-3}](https://latex.codecogs.com/gif.latex?\frac{10.8}{\left&space;[&space;207.2+\left&space;(&space;2\times&space;35.45&space;\right&space;)&space;\right&space;]}\;&space;mol\,&space;dm^{-3})
Solubility of Cl– with respect to Pb2+ = ![\frac{2\times 10.8}{\left [ 207.2+\left ( 2\times 35.45 \right ) \right ]}\; mol\, dm^{-3}](https://latex.codecogs.com/gif.latex?\frac{2\times&space;10.8}{\left&space;[&space;207.2+\left&space;(&space;2\times&space;35.45&space;\right&space;)&space;\right&space;]}\;&space;mol\,&space;dm^{-3})
![K_{sp}=\left [ Pb^{2+} \right ]\left [ Cl^- \right ]^2=\left ( \frac{10.8}{278.1} \right )\left ( \frac{2\times 10.8}{278.1} \right )^2=2.3\times 10^{-4}\; mol^{\: 3}\, dm^{-9}](https://latex.codecogs.com/gif.latex?K_{sp}=\left&space;[&space;Pb^{2+}&space;\right&space;]\left&space;[&space;Cl^-&space;\right&space;]^2=\left&space;(&space;\frac{10.8}{278.1}&space;\right&space;)\left&space;(&space;\frac{2\times&space;10.8}{278.1}&space;\right&space;)^2=2.3\times&space;10^{-4}\;&space;mol^{\:&space;3}\,&space;dm^{-9})
Question
Some solid Na2CO3 is slowly added to a solution containing 0.020 M of Zn2+ and 0.020 M of Ag+. Given that Ksp(ZnCO3) = 1.46 x 10-10 and Ksp(Ag2CO3) = 8.46 x 10-12, which cation will be precipitated first? What is the concentration of this cation in the solution when the other cation starts to precipitate?
Answer
[CO32-] required to begin Ag2CO3 precipitation is:

[CO32-] required to begin ZnCO3 precipitation is:

Therefore, Zn2+ will be precipitated first. When Ag2CO3 starts to precipitate,

Just as Ka and Kb are only useful for comparing weak acids and weak bases respectively, Ksp is only useful for comparing sparing soluble salts, as highly soluble salts have a higher probability of forming ion pairs. An ion pair consists of a cation and an anion that are electrostatically attracted to each other, rather than being individually surrounded by solvent molecules. This interaction alters their physical properties—for example, their mobility—and can distort the measurement of ion concentrations in solution when using ionic or conductivity methods. As a result, the accuracy of the Ksp value may be compromised. In general, the higher the solubility of a solid, the greater the concentration of ions in the solvent, which increases the likelihood of ion pair formation.
Furthermore, just as Kw is constant at a particular temperature regardless of the source of H3O+ and OH–, Ksp for a solid remains constant at a particular temperature regardless of the source of the dissolved ions. For example, the presence of NaCl in a saturated solution of AgCl causes the latter to precipitate, as Cl– is common to both species. The decrease in the solubility of a dissolved compound in the presence of an ion in common with the dissolved compound is called the common ion effect.
Question
Is Ksp dependent on the volume of the solution?
Answer
No. is a thermodynamic equilibrium constant that is governed by the formula
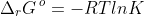
Hence, Ksp is only dependent on temperature. If we dilute a solution of PbCl2 that is in equilibrium with solid PbCl2, the increase in volume of the solution shifts the position of the equilibrium according to Le Chatelier’s principle to produce more aqueous Pb2+ and Cl– such that the saturation concentrations (mole per volume) of Pb2+ and Cl– remains unchanged when the new equilibrium is attained.
Another way to look at it is that Ksp is the mathematical product of the solubility of the ions of a compound, raised to the power of their respective stoichiometric coefficients in a particular volume of solvent at a particular temperature. Since solubility is an intensive property, Ksp is independent of the volume of solvent.