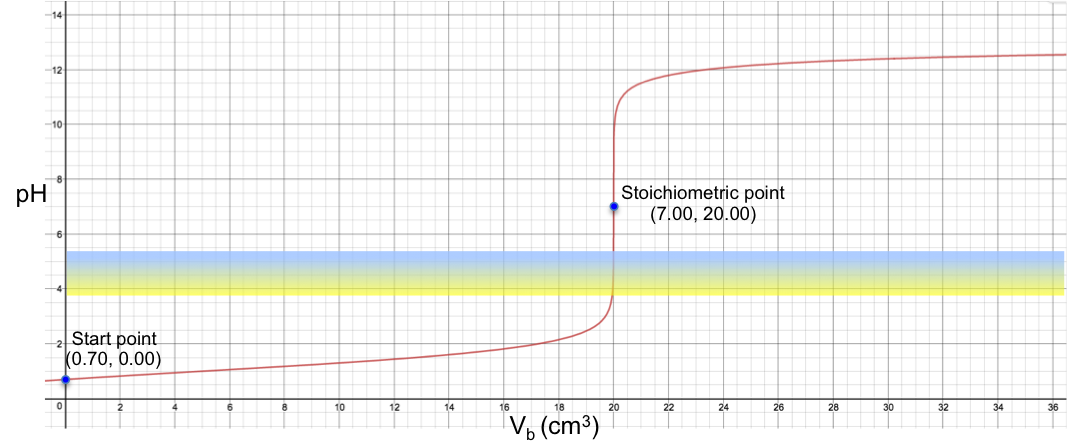
The effects of dilution on the pH of a strong acid (unbuffered solution) and a weak acidic buffer are shown in the diagram below.

It is evident from the diagram that the pH of the buffer solution is relatively resistant to dilution. However, as the solution is diluted infinitely, its pH will ultimately approach 7, the pH of water.
The pH of the systems are expressed by the following formulae:
|
System |
Approximate pH formula |
More accurate pH formula |
|
HCl |
————— |
|
| CH3COOH/CH3COO– buffer | ||
With reference to the above formulae, [H+] of a strong acid like HCl, decreases upon dilution and hence pH increases. According to the Henderson-Hasselbalch equation, the ratio of the concentration of the salt to the concentration of the acid is unchanged upon dilution and we would expect the buffer pH curve in the above diagram to be a horizontal line. However, the curve shows that the pH of a buffer solution appears relatively constant up to a thousand-fold dilution and subsequently increases at lower buffer concentrations. This is because the data points of the buffer curve in the diagram is calculated using the more accurate pH formula (i.e. the complete pH titration curve formula) instead of the Henderson-Hasselbalch equation, which only provides an approximation of the buffer’s pH. The Henderson-Hasselbalch equation is relatively accurate at high buffer concentrations or when Ka < 10-3, but unreliable at low buffer concentrations or when Ka > 10-3, where the assumptions used to derive it are no longer valid.


Question
Show that the resistance in the change in pH of the CH3COOH/CH3COO– buffer upon dilution is consistent with Le Chatelier’s principle.
Answer
The equilibrium constant for the reaction is given by:
Since Ka is a constant at a given temperature, the system must adjust the number of moles of each species upon dilution to respond to the increase in volume V. This is only possible if decreases, while
and
increase. Such an adjustment is consistent with Le Chatelier’s principle, which states that if a change disturbs a system at equilibrium, the system will shift in the direction that counteract the change. In this case, the system responds to the decreased concentrations of species upon dilution by shifting the equilibrium to the right, producing more moles of dissolved species. As a result,
decreases less than expected, demonstrating the buffer’s resistance to pH change.