Spin-spin coupling in nuclear magnetic resonance spectroscopy refers to the magnetic interactions between neighbouring nuclei that are not equivalent, leading to a change in resonance frequencies of the nuclei. As mentioned in an earlier article, the nucleus of an atom possesses a magnetic moment that generates a small magnetic field. If two NMR-active atoms (I ≠ 0) are in vicinity of each other, e.g. protons in the moiety HC-CH, the magnetic field of one 1H can shield or deshield the magnetic field of the other 1H, thereby slightly changing its shielding constant and therefore changing its resonance frequency.
In general, we refer to the following rules to determine the extent of nuclear spin-spin coupling:
-
- Spin-spin coupling occurs in all NMR-active nuclei. However, the coupling between two or more equivalent nuclei results in the same resonance energy of transition and therefore has no effect on the chemical shifts of the nuclei.
- Spin-spin coupling is negligible between nuclei separated by four or more bonds, e.g. the protons in HC-CH are separated by 3 bonds and consequently display coupling effects.
- The changes in chemical shifts of spin-coupled nuclei are usually much smaller than the difference in chemical shifts between the nuclei.
The effects of spin-spin coupling are visible on a high resolution NMR spectrum. An example is the spectrum of ethanol, CH3CH2OH:
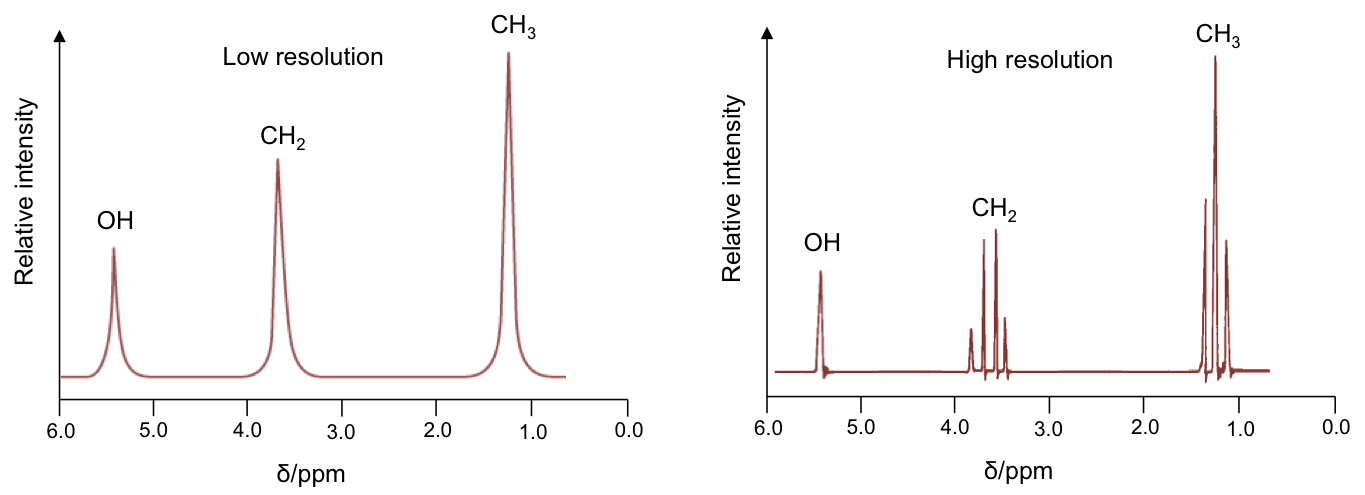
Effects of spin-spin coupling on CH3 protons
According to rule 1, the three 1Hs in the CH3 moiety are equivalent and spin-couple one another without affecting the position and shape of the CH3 peak in the spectrum. However, they are not equivalent to the two protons in CH2, which have the following possible spin orientations:
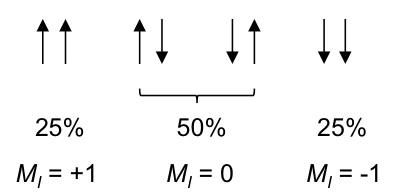
The percentages represent the statistical occurrences of the paired orientations in a sample of ethanol molecules, with MI = ΣmI , i.e. the total nuclear spin magnetic number. Depending on the spin orientation of a proton in the CH3 moiety, the paired orientations of MI = +1 or MI = -1 of protons in CH2 either shield or deshield that proton, while the paired orientations of MI = 0 in CH2 does not affect the proton’s magnetic field. The result is a splitting of the single CH3 peak into a triplet, with the middle peak retaining the same chemical shift as CH3 without spin-spin coupling effect, and an equal up and down shift for the left and right peaks. The relative intensity of the left:middle:right peaks is 1:2:1 (see high resolution diagram above).
CH3 protons are separated from the hydroxyl 1H by four bonds. So, according to rule 2, any coupling effect is negligible.
Effects of spin-spin coupling on CH2 protons
Using the same logic, the possible spin orientations of the three 1Hs in the CH3 moiety are:
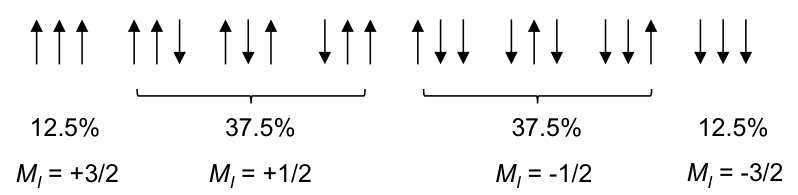
The four MI values split the single CH2 peak into a quartet, with the distribution of four peaks along a vertical line of reflection that intersects the original chemical shift of CH2 without spin-spin coupling effect. The relative intensity of the peaks is 1:3:3:1 (see high resolution diagram above).

Question
Why is the OH peak not a triplet and why is there no coupling effect between the hydroxyl 1H and the CH2 protons?
Answer
The presence of H3O+ or OH– in the sample, even in minute amounts, catalyses the exchange of hydroxyl protons between ethanol molecules. This exchange is fast enough to negate the spin-spin coupling effect between the hydroxyl proton and the CH2 protons. The hydroxyl protons can also be swapped with protons from H3O+, which is why the OH peak may disappear when D2O is added to the ethanol sample (ID = 0).
Finally, in cases where rule 3 fails, i.e. the changes in chemical shifts of spin-coupled nuclei are comparable to the difference in chemical shifts between the nuclei, the spectrum can become complex.
Question
Why are all peaks in a 13C NMR spectrum singlets?
Answer
13C, like 1H, has a nuclear spin of I = ½ and is expected to experience spin-spin coupling with neighbouring 13C and 1H nuclei. The natural abundance of 13C is only 1.1% and therefore the probability of a molecule having 13C adjacent nuclides is almost zero. However, 13C–1H spin-spin coupling is ubiquitous. To avoid a complicated 13C spectrum, a technique called proton decoupling is used, where protons of molecules in a sample are irradiated with a secondary radiofrequency wave. The protons undergo rapid spin reorientations that result in their net MI being zero.
Question
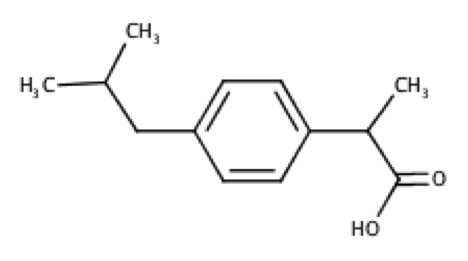
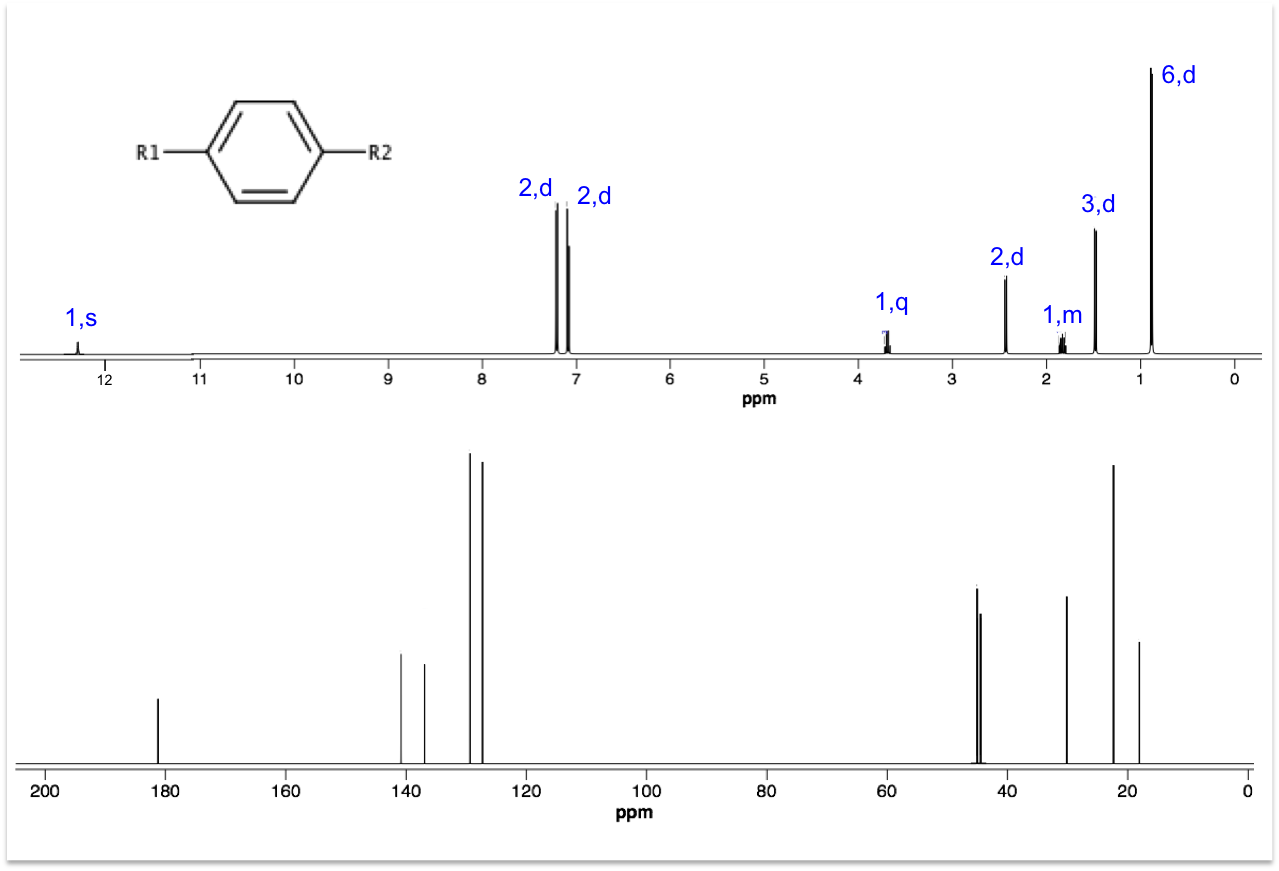
Deduce the structures for R1 and R2 using the 1H and 13C spectra below. Hint: Ar–13C 45 ppm.
Answer
Even though 13C peak areas are not a reliable indicator of the relative number of carbons, we can make a guess of the carbon numbers by comparing the peak heights. Let’s assume the three taller peaks represent two carbons each, which gives a total of 13 carbons in the molecule.
The peak at 0.90 ppm of the top spectrum is a doublet of 6 protons, which suggests an isopropyl group. The doublet of 3 protons at 1.50 ppm is mostly likely a methyl group. The singlet at 12.34 ppm is characteristic of a carboxyl proton. A pair of doublets at 7.08-7.22 ppm, each with two protons, refers to protons on the benzene ring. That leaves us with 2 carbons associated with the quartet at 3.69 ppm and the doublet at 2.45 ppm. These two carbons have almost the same chemical shift of 45 ppm in the 13C spectrum, which according to the hint are carbons adjacent to the benzene ring. Putting everything together, we have the following structure (ibuprofen):