The analytical solution of the energy equation of He involves finding analytical expressions for all terms in eq8 using trial one-electron wavefunctions. As per the numerical method, guess values are used for the variables in all trial one-electron wavefunctions except one, e.g. ) . Eq8 is then minimised to obtain the solutions for
. Eq8 is then minimised to obtain the solutions for 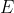 and . The process is repeated until and all become invariant. For He, eq8 is
and . The process is repeated until and all become invariant. For He, eq8 is
)
where \left(-\frac{1}{2}\nabla_i^2-\frac{Z}{r_i}\right)\phi_i(\mathbf r_i)d\mathbf r_i) and
and  .
.
Our computation employs the following assumptions:
- We base our iteration on the unrestricted case, where electrons
 and
and  are distinctively expressed by
are distinctively expressed by  and
and  respectively.
respectively.
- All terms on the RHS of eq48 are determined using Slater-type orbitals, where

Substituting in 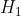 , using the identity
, using the identity  (see this article for proof) for
(see this article for proof) for  and integrating by parts for
and integrating by parts for  , we have
, we have
)
Similarly, substituting in 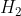 gives
gives
)
Substituting , and eq47 in  and multiplying the resultant equation by
and multiplying the resultant equation by ]^*Y_0^0(\theta_2,\phi_2)=1) , where
, where =\frac{1}{\sqrt{4\pi}}) is the spherical harmonics for
is the spherical harmonics for  , we have
, we have
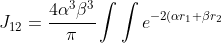}\sum_{n=0}^{\infty}\sum_{m=-n}^n\frac{4\pi}{2n+1}\frac{r_<^n}{r_>^{n+1}})
[Y_n^{\vert m\vert}(\theta_2,\phi_2)]^*[Y_0^0(\theta_1,\phi_1)]^*Y_0^0(\theta_2,\phi_2)d\boldsymbol{\mathit{r}}_1d\boldsymbol{\mathit{r}}_2)
Due to the orthogonality of the spherical harmonics, the only integral that survives upon expanding the summation is when  and
and 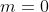 . Since,
. Since,  , the above equation becomes:
, the above equation becomes:

We proceed by integrating with respect to 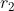 first. Since ranges from 0 to
first. Since ranges from 0 to 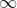 , we can split the integral into two parts, one from 0 to
, we can split the integral into two parts, one from 0 to 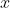 and the other from to . Supposing
and the other from to . Supposing  , the above equation becomes:
, the above equation becomes:
dr_1)
As increases from 0 and approaches 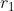 , it must be less than and so
, it must be less than and so  . Similarly, as increases from to , it must be greater than and so
. Similarly, as increases from to , it must be greater than and so  . Therefore,
. Therefore,

dr_1\;\;\;\;\;\;\;\;(52))
Using the identity ^i}{i!}\right]) for the first integral within parentheses, integrating by parts for the second integral within parentheses, and employing the identity
for the first integral within parentheses, integrating by parts for the second integral within parentheses, and employing the identity  for the integral with respect to yields
for the integral with respect to yields
}{(\alpha+\beta)^3}\;\;\;\;\;\;\;\;55)
Substituting eq50, eq51 and eq55 in eq48 gives
}{(\alpha+\beta)^3}\;\;\;\;\;\;\;\;56)
Differentiating eq56 with respect to 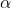 and
and 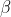 results in
results in
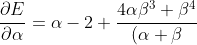^4}\;\;\;\;\;\;\;\;57)
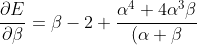^4}\;\;\;\;\;\;\;\;58)
Eq57 and eq58 are used in an iterative algorithm to find , and . The procedure is as follows:
- Substitute an initial guess value of , e.g.,
 , in eq57 and solve for by setting eq57 equal to zero. We then substitute the solution of and the initial guess value of in eq56 to find .
, in eq57 and solve for by setting eq57 equal to zero. We then substitute the solution of and the initial guess value of in eq56 to find .
- To obtain an improved estimate of , we substitute the solution of from the previous step in eq58, and solve for by setting eq58 equal to zero. We then substitute the solution of and the value of found in the previous step in eq56 to find a better estimate of .
- Steps 1 and 2, which form an iteration set, are repeated until the values of and are invariant up to six decimal points.
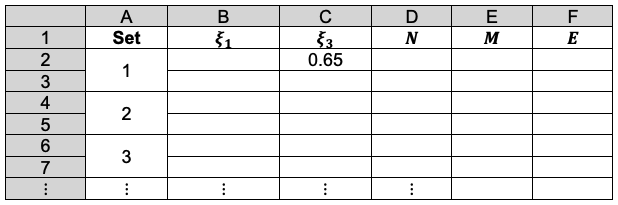
Alternatively, we can set up an iterative table in Excel as follows:

Cell C2 is the initial guess value of . The formula for D2:D7 is eq56, i.e. =(B2^2)/2-2*B2+(C2^2)/2-2*C2+(B2*C2*((B2^2)+3*B2*C2+(C2^2)))/((B2+C2)^3). The formulae for B3, C4, B5, C6 and B7 are =B2, =C3, =B4, =C5 and =B6, respectively. To compute the first iteration set, we employ the Excel Solver application to minimise D2 with respect to B2, with the following settings:
Set objective: D2
To: Min
By changing variable cells: B2
Selecting a solving method: GRG Nonlinear
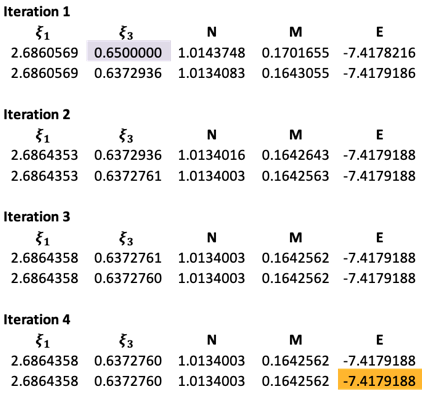
We then solve for D3 by changing the ‘Set objective’ field and ‘By changing variable cells’ field to D3 and C3 respectively. This procedure is repeated for subsequent iteration sets until the values of and are invariant up to six decimal points.
The results are as follows:

The final theoretical value of  has a deviation of about 1.93% versus the experiment data of the ground state of helium.
has a deviation of about 1.93% versus the experiment data of the ground state of helium.