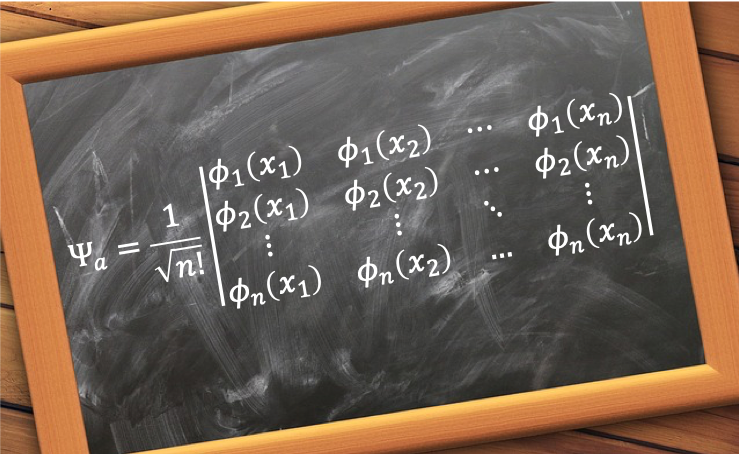To solve the Hartree-Fock-Roothaan equations for He, we begin by noting that in the spin orbitals
and
. If the basis wavefunctions
and
are real,
becomes
. With reference to eq157, we have
where (assuming no contribution from the exchange integral) and
.
The pair of simultaneous equations can be represented by the following matrix equation:
Let the basis wavefunctions be and
. Since these Slater-type orbitals are real,
. Eq158 becomes

Therefore, eq159 becomes
Eq160 is a linear homogeneous equation, which has non-trivial solutions if
Expanding the determinant and noting that , we get the characteristic equation:
The computation process is as follows:
- Evaluate the integrals
,
,
and
in eq161 either analytically or numerically by letting
and
, and using the initial guess values of
.
- Substitute the evaluated integrals in eq161, solve for
and retain the lower root. Substitute
in terms of
.
- Use the expression derived in part 2, together with
, to solve for
and
for the next iteration.
- Repeat steps 1 through 4 until
The results are as follows: