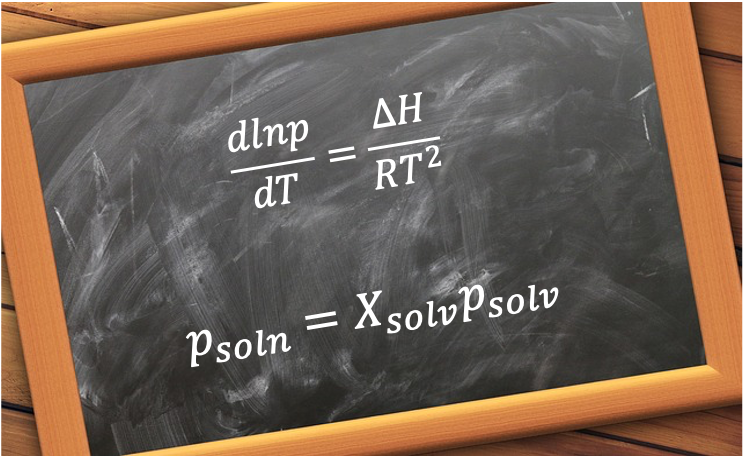A permutation of a set is an ordered arrangement of its elements.
Distinct elements
Consider the task of filling  buckets, each with a ball chosen from a pool of distinct numbered balls. There are possible choices for the first bucket. Once the first ball is used, only
buckets, each with a ball chosen from a pool of distinct numbered balls. There are possible choices for the first bucket. Once the first ball is used, only 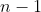 choices remain for the second bucket, then
choices remain for the second bucket, then 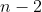 choices for the third bucket, and so on. This continues until the last bucket, for which only 1 ball remains.
choices for the third bucket, and so on. This continues until the last bucket, for which only 1 ball remains.
If you have 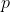 ways to do a first task, and for each of those,
ways to do a first task, and for each of those, 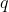 ways to do a second task, then the total number of ways to perform both tasks is
ways to do a second task, then the total number of ways to perform both tasks is  . This principle extends to any number of sequential tasks. Therefore, the total number of distinct arrangements (permutations) of the balls in buckets, denoted by
. This principle extends to any number of sequential tasks. Therefore, the total number of distinct arrangements (permutations) of the balls in buckets, denoted by ) or
or  , is the product of the number of choices for each task:
, is the product of the number of choices for each task:
=n(n-1)(n-2)\cdots(2)(1)=n!)
Now, suppose there are distinct balls but only  buckets, where
buckets, where  . Again, there are choices for the first bucket, for the second, and so on until the -th bucket. Since we can also express the number of choices for the second bucket as
. Again, there are choices for the first bucket, for the second, and so on until the -th bucket. Since we can also express the number of choices for the second bucket as  , we have
, we have  ways of filling the -th bucket. Hence,
ways of filling the -th bucket. Hence,
=n(n-1)(n-2)\cdots(n-r+1)\;\;\;\;\;\;\;\;306)
Multiplying RHS of eq306 by !}{(n-r)!}) gives
gives
=n(n-1)(n-2)\cdots(n-r+1)\frac{(n-r)!}{(n-r)!}=\frac{n!}{(n-r)!}\;\;\;\;\;\;\;\;307)
Eq307 gives the general formula for calculating permutations — the number of ways to arrange objects selected from a set of distinct objects.

Question
In a lottery draw for a four-digit winning number, each digit can be any number from zero to nine. How many permutations are possible?
Answer
Total permutations = 10 x 10 x 10 x 10 = 104. In general, the number of permutations for forming a sequence of positions, where each position can be filled with possible choices is  .
.
Repeating elements (multinomial permutation)
How many distinct permutations are there of the letters in the word “MISSISSIPPI”? If all letters were distinct, the number of permutations would be 11! = 39,916,800. However, some letters are repeated in the word: 4 I’s, 4 S’s, 2 P’s and 1 M. To understand why we need to adjust for repeated letters, let’s consider the I’s. Suppose we label the four I’s as I1, I2, I3 and I4, treating them as distinct. In any arrangement, these four labelled I’s can be rearranged among themselves in 4! = 24 ways. For example, we could have:
M I1 S S I2 S S I3 P P I4
M I1 S S I2 S S I4 P P I3
M I2 S S I1 S S I3 P P I4
…and so on.
But since these I’s are actually indistinguishable, all 24 of those arrangements represent the same permutation of the word. So, we’ve overcounted each unique arrangement by a factor of 4!. Similarly, the 4 S’s and 2 P’s can be rearranged in 4! ways and 2! ways respectively. Each of these reorderings also causes overcounting, resulting in a total overcounting factor of 4! x 4! x 2!. Therefore, to find the number of distinct permutations of the letters in “MISSISSIPPI”, we divide the total number of arrangements by the total repeated counts:  .
.
In general, if you have a set of objects, where 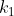 are identical to each other,
are identical to each other, 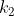 are identical to each other but different from the first group, and so on until
are identical to each other but different from the first group, and so on until 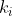 , each representing a distinct group of identical items, then the number of distinct permutations is:
, each representing a distinct group of identical items, then the number of distinct permutations is:

where  and
and  is also known as the multinomial coefficient.
is also known as the multinomial coefficient.
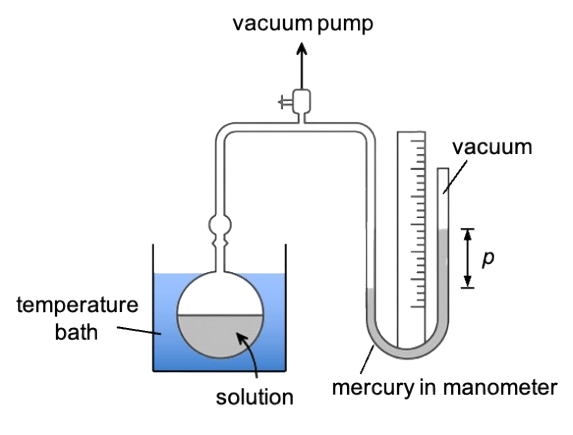
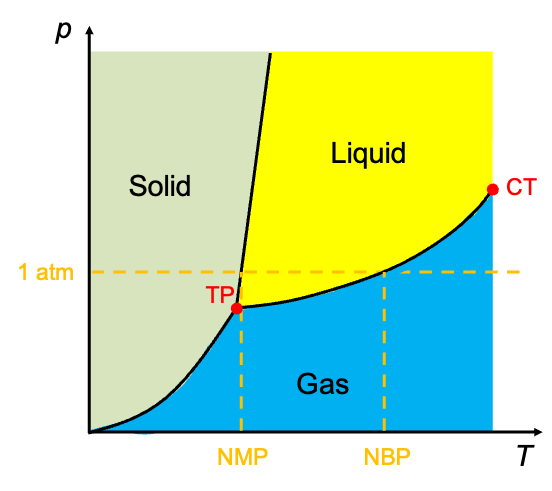
Eq308 is used to derive the Boltzmann distribution and Raoult’s law.
A combination is a selection of items where the order does not matter. For example, AB and BA are two different permutations of the set {A,B}, but they represent the same combination. To calculate the number of combinations, we start by counting the permutations (where order does matter), and then correct for overcounting by dividing out the number of ways the selected items can be rearranged.
Suppose we are selecting objects from a set of distinct objects. The number of permutations is given by eq307: !}) . However, items can be arranged in
. However, items can be arranged in  ways, all of which count as the same combination when order doesn’t matter. There, the number of combinations, denoted by
ways, all of which count as the same combination when order doesn’t matter. There, the number of combinations, denoted by ) or
or  or
or ) , is
, is
=\frac{n!}{r!(n-r)!}\;\;\;\;\;\;\;\;309)
Interesting, the number of combinations is also the binomial coefficient.
Question
Show that a mutlinomial permutation can be expressed as a product of combinations.
Answer
Suppose we want to partition a set of distinct items into 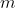 groups of specified sizes , , …,
groups of specified sizes , , …, 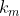 , where
, where  . The number of ways to select items for the first group is
. The number of ways to select items for the first group is
=\frac{n!}{k_1!(n-k_1)!})
The number of ways to select the remaining  items for the second group is
items for the second group is
=\frac{(n-k_1)!}{k_2!(n-k_1-k_2)!})
The number of ways to select the remaining  items for the third group is
items for the third group is
=\frac{(n-k_1-k_2)!}{k_3!(n-k_1-k_2-k_3)!})
and so on until the remaining the number of ways to select the remaining items for the last group is

Therefore, the total number of ways to perform all the sequential tasks is
!}\frac{(n-k_1)!}{k_2!(n-k_1-k_2)!}\frac{(n-k_1-k_2)!}{k_3!(n-k_1-k_2-k_3)!}\cdots\frac{k_m!}{k_m!0!})
which simplifies to the RHS of eq308 after all the cancellations.