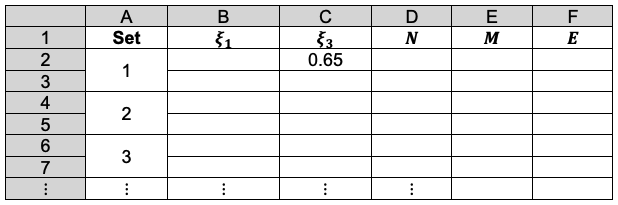
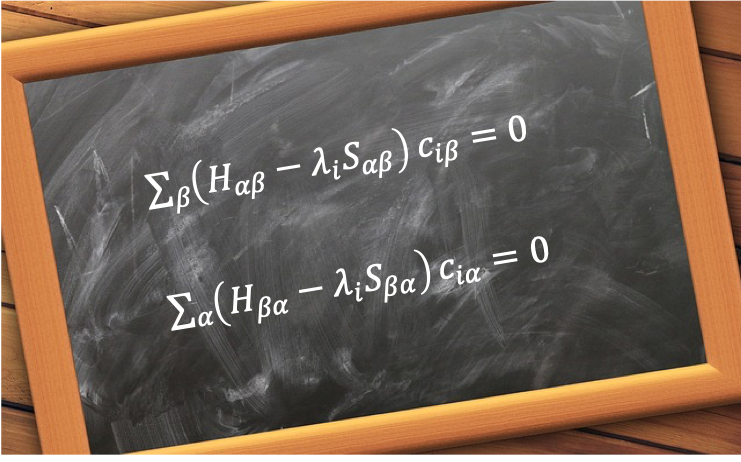The Gram-Schmidt process is a mathematical technique for orthogonalising a set of nonorthogonal vectors.
Consider two linearly independent nonorthogonal vectors and
of the Hermitian operator
:
where is the degenerate eigenvalue corresponding to
and
.

Let . With reference to the diagram above,
, where we have used the fact that the magnitude of the unit vector
is 1 in the second equality. Furthermore,
; that is, the vector
is a multiple of the unit vector
, with the multiple being
. Therefore,
is a component of
, which is orthogonal to
. The eigenvalue of
is unchanged versus that of
because
In the presence of a third linearly independent vector that is nonorthogonal to
and
(where
), the vector
that is orthogonal to
is
(c.f. eq111). To determine the component of
that is orthogonal to both
and
, let
be the component of
that is orthogonal to
:
We can immediately see that is orthogonal to both
and
because it is sum of two vectors
and
, each of which is orthogonal to
(and hence the dot product
is zero). Substituting
in the above equation, noting that
and
are scalars and that
, we have
Therefore, for a set of three linearly independent nonorthogonal vectors , the transformed set of vectors, which are orthogonal to one another, is
, where
For a set of linearly independent nonorthogonal vectors , the transformed set of vectors, which are orthogonal to one another, is
with the k-th transformed vector as
The corresponding orthonormal vectors are ,
, … ,
.
An example of the application of the Gram-Schmidt process is the orthogonalisation of nonorthogonal Slater-type orbitals in the Hartree-Fock method.